
 Inhalt Index
Inhalt IndexAufgrund der starken Aromas, das viele Benzolderivate ausströmen, nennt man sie aromatische Verbindungen. Deshalb gilt Benzol als Stammverbindung der Aromaten. Aromatische Ringe kommen in vielen biologisch wirksamen Substanzen vor.
z.B.:
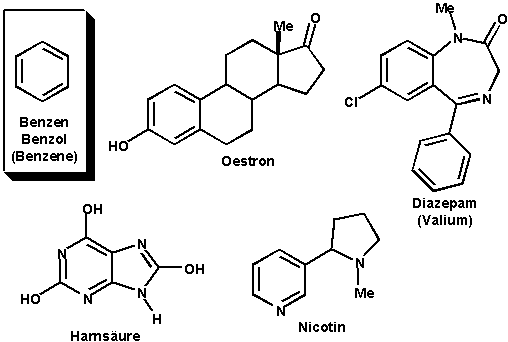
Wir werden hier nur Benzol-Derivate betrachten, und nicht aromatische Ringe, die ein Heteroatom enthalten (Kapitel 6), z.B. N, O oder S.
Der allgemeine Ausdruck für substituierte Benzolderivate lautet Arene. Ein Aren als Substituent heisst Arylgruppe, abgekürzt Ar-. Die einfachste Arylgruppe heisst Phenyl-, C6H5-, abgekürzt Ph-.
Einige Trivialnamen die häufig vorkommen:
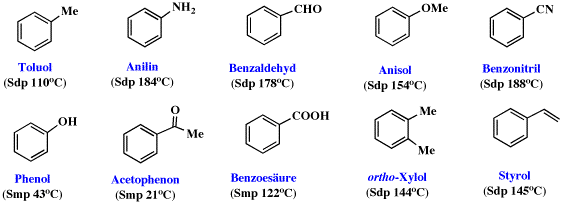
Viele monosubstituierte Benzolderivate benennt man, indem man den Substituentennamen dem Wort "Benzol" voranstellt :
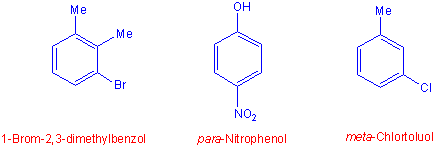
Bei disubstituierten Benzolderivaten können die Substituenten drei mögliche Stellungen zueinander einnehmen: benachbarte Substituenten 1,2- (ortho), für 1,3-disubstituierte Verbindungen 1,3- (meta), für 1,4-disubstituierte Verbindungen 1,4- (para). Die Substituenten werden in alphabetischer Reihenfolge genannt.
Im Jahr 1825 erhielt der englische Wissenschaftler Faraday eine farblose Flüssigkeit mit der empirischen Formel CH. Diese Verbindung war mit der Theorie, nach der jeder Kohlenstoff vier Valenzen zu anderen Atomen ausbilden musste, nicht in Einklang zu bringen. Besonders die ungewöhnliche Stabilität und chemische Trägheit dieser Substanz fiel auf. Man nannte die Verbindung Benzol und stellte schliesslich die Summenformel C6H6 dafür auf.
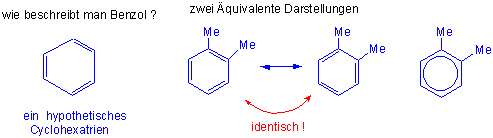
Ein Problem damals war, zu verstehen, warum nie zwei unterschiedliche Isomere von einem 1,2-disubstituierten Benzolderivat beobachtet werden konnten. Als Lösung schlug Kekulé im Jahre 1865 vor, dass Benzol aus sich rasch ineinander umwandelnden Isomeren von Cyclohexatrien bestehen sollte. Heute wissen wir, dasss diese Hypothese nicht völlig richtig ist. Nach der modernen Elektronentheorie kann man Benzol durch zwei äquivalente Resonanzstrukturen des Cyclohexatriens beschreiben:
Benzol ist ungewöhnlich reaktionsträge. Es geht keine Additionsreaktionen wie normale Alkene ein.
Molekülorbital-Modell des Benzols
Die Abbildung zeigt uns die elektronische Struktur des Benzolrings. Alle Kohlenstoffatome sind sp2-hybridisiert und jedes p-Orbital überlappt gleichmässig mit seinen beiden Nachbarn. Die auf diese Weise delokalisierten Elektronen bilden eine π-Elektronen-Wolke oberhalb und unterhalb der Ringebene.
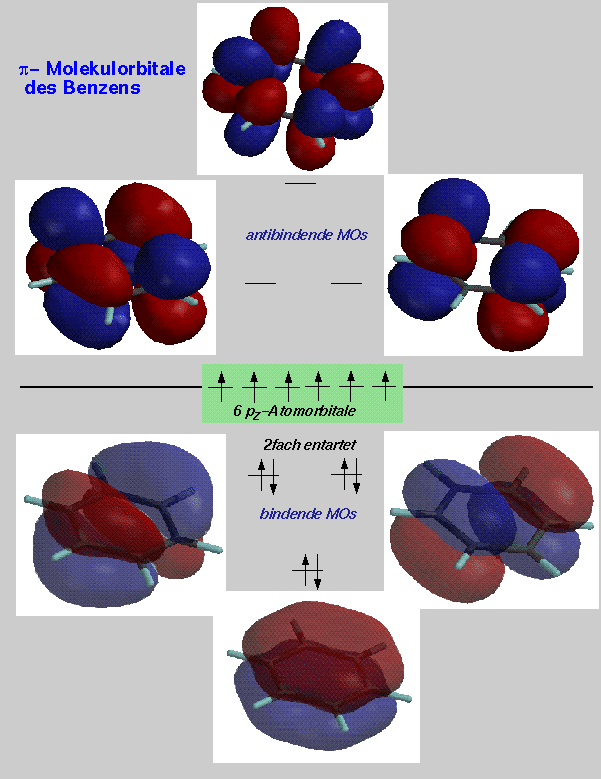
Die sechs cyclisch angeordneten, überlappenden p-Orbitale bilden einen Satz von sechs Molekülarorbitalen.
Das Benzolmolekül bildet ein reglmässiges Sechseck aus sechs sp2-hybridisierten Kohlenstoffatomen. Die Länge der aromatischen C-C-Bindung liegt zwischen der einer Einfach- und der einer Doppelbindung.
Valenz-Struktur-Beschreibung des Benzols
Die Struktur von Benzol kann durch zwei gleichwertige Resonanzstrukturen von Cyclohexatrien
wiedergegeben werden:
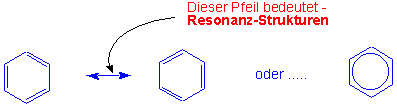
Der wirkliche Zustand wird dann als "Zwischenzustand " oder "Resonanz-hybrid" zwischen diesen Grenzstrukturen wiedergegeben.
Das Zeichen <-> bedeutet, dass nicht etwa ein dynamisches Gleichgewicht zwischen zwei verschiedenen Molekülarten existiert, sondern dass der tatsächliche Zustand zwischen den Grenzstrukturen liegt und von diesen gewissermassen "umschrieben wird". Die Beschreibung einer wirklichen Struktur durch Kombination von nicht existierender Grenzstrukturen nennt man Resonanz.
Um ein Mass für die relative Stabilität einer Reihe von Alkenen zu bekommen, kann man ihre Hydrierungswärmen bestimmen. Ein ähnliches Experiment können wir mit Benzol durchführen, und seine Hydrierungswärme mit der von l,3 Cyclohexadien und Cyclohexen vergleichen :
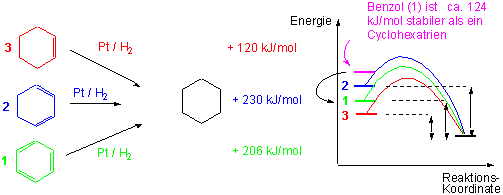
Obwohl Benzol nur schwer hydriert wird, kann die Reaktion katalytisch durchgeführt werden , und man erhält für die Hydrierungswärme einen Wert von ΔHo-206 kJ/mol. Der Unterschied zwischen den Hydrierungswärmen (und den Bildungsenthalpien ) beträgt ungefähr 124 kJ/mol und wird als Resonanzenergie von Benzol bezeichnet. Andere Namen dafür sind Delokalisierungsenergie, aromatische Stabilisierug, oder einfach Aromatizität von Benzol.
Mit Hilfe quantentheoretischer Rechnungen lässt sich zeigen, dass ein monocyclisches konjugiertes Polyen besonders stabil ist (d.h. Aromatizität besitzt), wenn es 4n+2 π-Elektronen enthält (n ist ganzzahlig), z.B.:
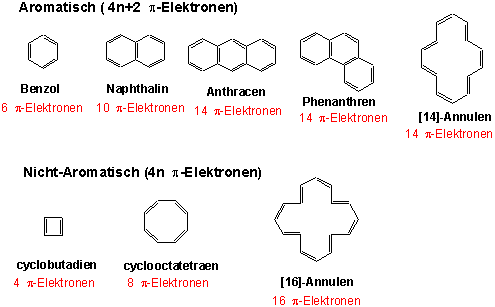
Aufgrund seiner π-Elektronen besitzt das Benzol-Molekül nucleophile Eigenschaften und reagiert deshalb vorwiegend mit Elektrophilen. z.B.:
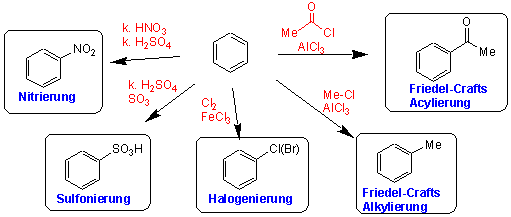
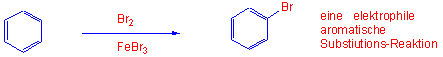
Im Gegensatz zu den Alkenen führt ein solcher Angriff zu einer Substitution eines H-Atoms, nicht zu einer Addition. z.B. Bromierung in Gegenwart eines Katalysators:
Mechanismus der elektrophilen aromatischen Substitution:
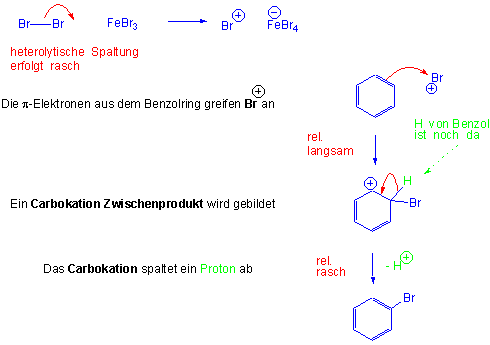
Der erste Schritt ist thermodynamisch ungünstig, da die cyclische Delokalisierung und damit der aromatische Charakter dabei verloren geht. Nach diesem Schritt wird der aromatische Ring wieder regeneriert, indem das Proton von dem sp3-hybridisierten Kohlenstoff abgespalten wird. Dies ist energetisch günstiger als eine Reaktion mit einem Nukleophil, wodurch das Additionsprodukt entstünde :
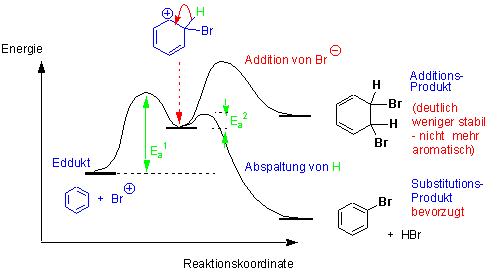
Die Änderung der potentiellen Energie während der Reaktion von Benzol mit einem Elektrophil. Die Bildung des ersten Übergangszustand ist geschwindigkeitsbestimmend. Das Proton wird verhältnismässig rasch abgespalten.
Die Reaktion von Benzol mit konz. HNO3 bei mässig erhöhter Temperatur führt zur Nitrierung des Benzolrings:
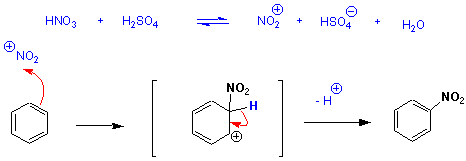
Konz. H2SO4 reagiert bei RT nicht mit Benzol, sieht man von der Protonierung ab. "Rauchende Schwefelsäure" (Oleum) greift jedoch elektrophil an, da sie SO3 enthält. Aufgrund der stark elektronenziehenden Wirkung der drei Sauerstoffatome ist das S-Atom in SO3 so elektrophil, dass es Benzol direkt angreift :
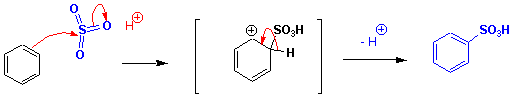
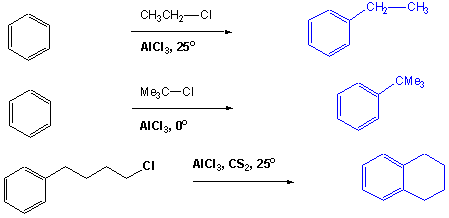
In Friedel-Crafts-Reaktionen entstehen neue C-C Bindungen. In Gegenwart einer Lewis Säure, gewöhnlich Aluminiumchlorid, greifen Halogenalkane Benzol unter Bildung von Alkylbenzolderivaten an (Friedel-Crafts Alkylierung), Alkanoylhalogenide ergeben Alkanoylderivate (Friedel-Crafts Acylierung):


Die Reaktivität des Halogenalkans nimmt in der Reihenfolge RF > RCl > RBr > RI ab. Typische Lewis-Säuren sind bei dieser Reaktion (nach abnehmender Reaktivität geordnet) AlBr3, AlCl3, FeCl3, SbCl5 und BF3. Weitere Beispiele:
Durch Friedel-Crafts-Acylierung entstehen Aryl-Alkyl-Ketone. z.B. Benzol reagiert mit Acetylchlorid in Gegenwart von AlCl3 und bildet Acetophenon :
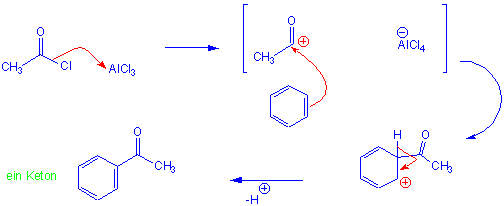
Die Bildung des Acylium-Kations geschieht allgemein durch die Reaktion von Alkanoylhalogeniden (Acylhaliden) mit AlCl3. Ähnlich reagieren Carbonsäureanhydride mit Lewis-Säuren.
z.B.:
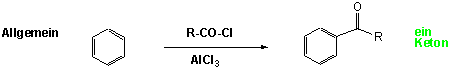
Ein Substituent am Benzolring übt einen elektronischen Effekt aus, indem er entweder Elektronendichte auf den Ring überträgt, oder Elektronendichte abzieht. Auf diese Weise kann ein monosubstituierter Benzolring, verglichen mit Benzol, andere chemische und physikalische Eigenschaften aufweisen, z.B. kann eine elektrophile aromatische Substitutionsreaktion viel langsamer (d.h. wird desaktiviert) oder schneller (d.h. wird aktiviert) ablaufen als mit Benzol. Das Abziehen oder Liefern von Elektronen kann durch Induktive- und durch Resonanz-Effekte zustandekommen.
Induktive- und Resonanz-Effekte beeinflussen viele chemische und physikalische Eigenschaften von Aromaten, nicht nur Ihre Reaktivität bei elektrophilen Substitutionsreaktionen.
Induktive- und Resonanz-Effekte
Betrachten wir Beispiele von elektronenliefernden und elektronenziehenden Substituenten:
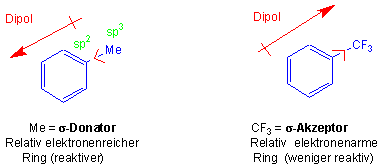
Induktive Effekte sind Polarisationseffekte, die über σ-Bindungen übertragen werden (siehe Kapitel 1.10).
Es ist auch möglich, dass ein Substituent an einen aromatischen Ring mit p-Elektronen zu den p-Elektronen des Ringes in Konjugation tritt und dadurch entweder negative Ladung aus dem ungesättigten System abzieht oder negative Ladung in dieses hineindrückt. Häufig spricht man von einem mesomeren Effekt oder Resonanzeffekt, was daherrührt, dass man in diesen Fällen die Ladungsdichteverteilung durch Kombination verschiedener Grenzstrukturen beschreiben kann, da eine Delokalisation der p-Elektronen eintritt. z.B. ein π-Akzeptor
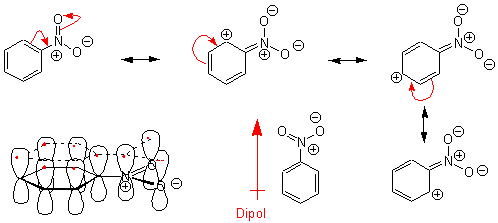
Ihre Akzeptorwirkung (die Stärke des mesomeren Effektes) steigt mit der Bereitschaft des Substituenten, negative Ladung aufzunehmen.
Andere π-Akzeptorengruppen: In der folgenden Reihe nimmt er darum nach rechts zu:
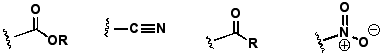
π-Donorgruppen: Da N elektronegativer ist als C, übt die -NH Gruppe2 einen leicht elektronenziehenden Effekt aus (induktiver Effekt). Das freie Elektronenpaar des N-Atoms kann jedoch in das aromatische π-System miteingebracht werden, sodass die Ladungsdichte des Ringes erhöht wird:

Dieser Resonanzeffekt überwiegt den induktiven Effekt bei weitem. Anilin ist deshalb für weitere Substitution leicht zugänglich.
Das elektronegative O-Atom in Phenol wirkt ebenfalls elektronenziehend (induktiver Effekt). Aber auch hier überwiegt der Einfluss der Resonanz (p-Donoreffekt), sodass der Benzolkern elektronenreicher wird:
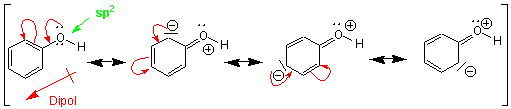
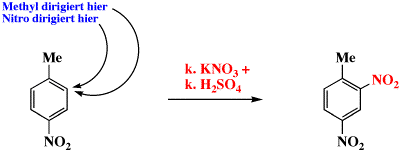
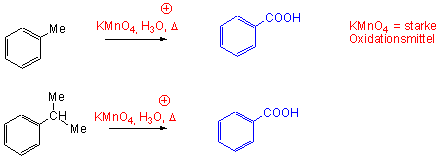
Übt ein bereits vorhandener Substituent einen Einfluss darauf aus, an welcher Stelle im Ring ein Elektrophil angreift, und wie schnell die reaktion abläuft (im Vergelich zum Benzol)? Schauen wir zuerst Toluol an, das die induktiv aktivierende Methylgruppe trägt:
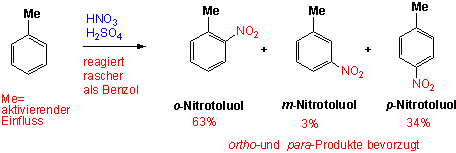
Die elektrophile Nitrierung von Toluol führt hauptsächlich zu ortho und para-Substitution.
Die Nitrierung ist kein Spezialfall:
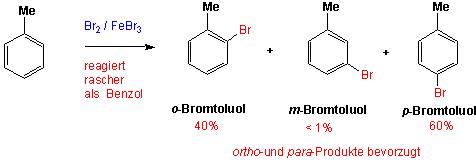
Trifluormethylbenzol ist gegenüber dem elektrophilen Angriff desaktiviert und reagiert deshalb nur zögernd. Unter energischen Bedingungen aber erhält man Substitution, jedoch nur in meta-Stellung:
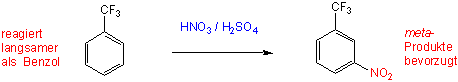
Für einen elektrophilen Angriff auf einem resonanz-aktivierten Benzolring ist oft nicht einmal ein Katalysator notwendig. Die Reaktion verläuft rasch und vollständig regioselektiv zu den (oft mehrfach) substituierten ortho- und para-Produkten:
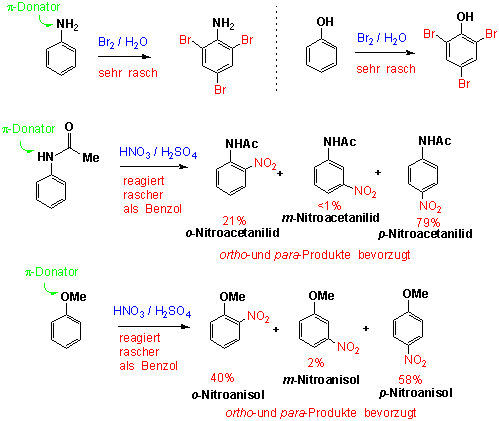
Anisol reagiert ungefähr 1000x schneller als Benzol! Wenden wir uns nun Benzolderivaten zu, die durch Resonanz desaktivierende Gruppen tragen. Hierher gehört die Benzoesäure, bei der die Nitrierung 1000x langsamer verläuft als bei Benzol:
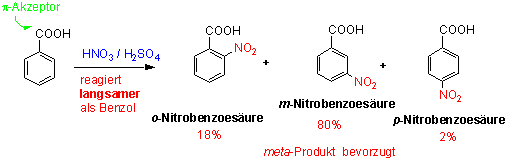
Die Nitrierung führt überwiegend zum meta-Produkt.
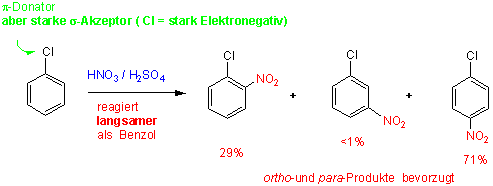
Die Halogene (-F, -Cl, -Br, -I) als Substituenten bilden eine dritte Gruppe, die den Ring desaktivieren (Chlorbenzol reagiert ca. 15-fach langsamer als Benzol), jedoch hauptsächlich ortho- und para- substituierte Produkte bilden :
Dirigierende Wirkung von Substituenten bei der elektrophilen aromatischen Substitution:
| Ortho- und para-dirigierend | Ortho- und para-dirigierend | Meta-dirigierend
| aktivierend | desaktivierend | desaktivierend
| (σ und π-Donorgruppen) | | (σ und π-Akzeptorgruppen)
| -NH2, -NHR, -NR2 | -F, -Cl, -Br, -I | -NO2 -CF3
| -NH-COR | | -NR3+, -COOH
| -OH, -OR | | -COOR, -CO-R
| -R (Alkyl, Aryl) | | -SO3H, -CN
| |
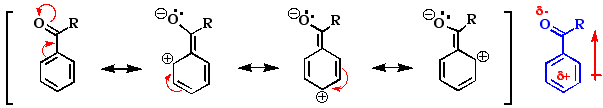
Können wir Mechanismen aufstellen, die diese Selektivitäten erklären können ? Dazu wollen wir die möglichen Resonanzstrukturen des Kations zeichnen, das nach dem Angriff des Elektrophils (E+) entsteht. Dabei stellen wir fest, dass σ und π-Donorsubstituenten, die in ortho oder para Stellung stehen, das Zwischenprodukt (und den dazu führenden Übergangszustand) stabilisieren:

Nur durch den Angriff in ortho- und para-Stellung entsteht ein Kation mit einer Resonanzstruktur, die eine positiven Ladung neben der Alkylgruppe trägt. Da diese Struktur etwas vom Charakter eines 3o-Carbenium-Ions hat, kommt ihr mehr Bedeutung zu als anderen, die die positive Ladung an einem 2o C-Atom tragen.
Durch einen meta-Angriff dagegen entsteht eine Zwischenstufe, in der keine der möglichen Resonanzstrukturen von der Stabilität eines 3o-Carbenium-Ions profitiert. Der elektrophile Angriff auf ein C-Atom ortho- oder para- zu einer Methyl(Alkyl)-Gruppe führt deshalb zu einer Zwischenstufe, die stabiler ist als die, die durch einen meta-Angriff entstehen würde. Sie entsteht daher relativ schnell über einen Übergangszustand mit relativ niedriger Energie (Vgl. Kapitel 5.5).
Auch beim elektrophilen Angriff auf einen Resonanz-aktivierten Benzol-Ring kann man die beobachtete Regioselektivitäten durch Resonanzstrukturen der verschiedenen intermediären Kationen erklären, z.B.:
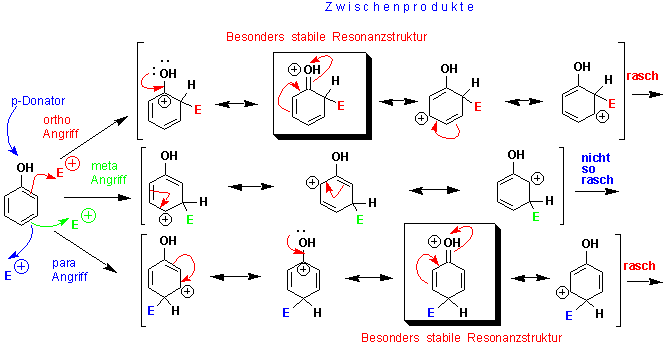
Ortho- und para-Substitutionen sind bevorzugt, da sie über Kationen verlaufen, für die man vier Resonanzstrukturen aufstellen kann, während bei der Zwischenstufe eines meta-Angriffs nur drei zu formulieren sind.
Wenn wir nun Benzolderivate betrachten, die Resonanz desaktivierende Gruppen tragen:
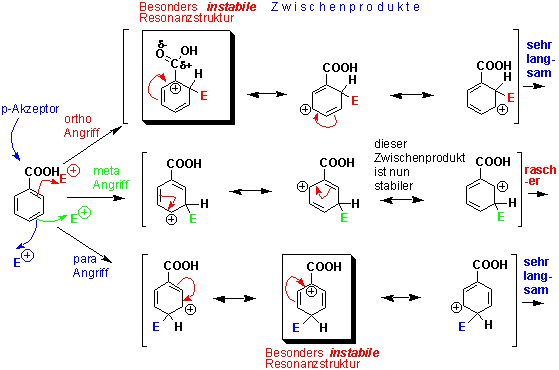
Der Angriff auf die meta-Position vermeidet hier, dass eine positive Ladung neben der elektronenziehenden Carboxygruppe entsteht. Bei ortho- und para- Angriff hingegen müssen recht energiereiche Resonanzstrukturen formuliert werden. Bei anderen desaktivierenden Substituenten ist die Situation analog.
Halogenatome wie Cl und Br sind räumlich viel grösser als O- und N-Atome. Obwohl ein p-Donoreffekt von -Cl noch vorhanden ist, ist der Resonanzeffekt nicht so wirksam (wie bei N- und O-Atomen), weil die p-Orbitale am -Cl schlechter mit den benachbarten p-Orbitalen des Benzolringes überlappen. N-, O- und Cl-Atome sind alle elektronegativ (σ-Akzeptorwirkung), aber mit O- und N-Atomen ist der Resonanzeffekt (p-Donor) viel stärker. Das heisst, Chlorbenzol ist weniger reaktiv, bevorzugt aber immer noch Substitutionsreaktionen in ortho- und para-Positionen. z.B.
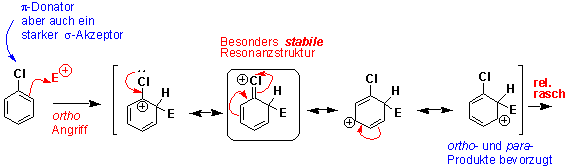
Der Resonanzeffekt führt ebenfalls zu ortho- und para-Produkten.
Die Wirkungen zweier Substituenten auf die relative Geschwindigkeit und auf die Orientierung der elektrophilen Substitution des Benzolrings addieren sich, z.B.:
Wenn zwei in entgegengesetzte Richtung lenkende Gruppen vorhanden sind, sie üben ihren Einfluss gewöhnlich unabhängig voneinander aus. Konkurrieren die Substituenten miteinander um den Ort der Substitution, setzt sich die stärker aktivierende (oder desaktivierende) Gruppe durch. Resonanz-Effekte sind normalerweise stärker als Induktive-Effekte.
z.B.:
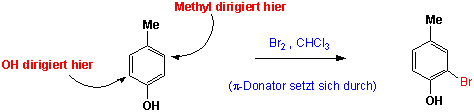
Wenn räumlich ausgedehnte Gruppen vorhanden sind, ist eine Stellung zwischen diesen Substituenten oft aus sterischen Gründen ungünstig:
z.B.
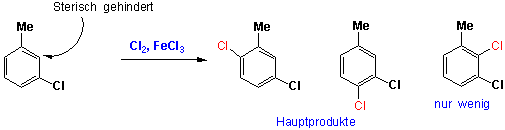
In den meisten anderen Fällen ergeben sich Produktgemische.
Benzol fällt bei verschiedenen technischen Prozessen an, z.B. in der aromatischen Fraktion von Rohöl-Destillat. Jährlich werden weltweit etwa 35 Millionen Tonnen Benzol hergestellt. Von 1940 bis ungefähr 1960 wurde das meiste Benzol auf der Basis der Steinkohle hergestellt. Seit 1950 wird es auch aus Erdöl gecrackt.
Syntheseplanung
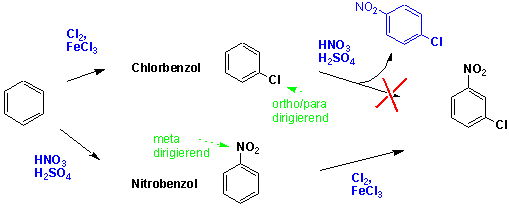
Bei der Synthese eines spezifischen Benzolderivats hängt alles davon ab, ob der erste eingeführte Substituent weitere Substituenten in die richtige Position dirigiert:
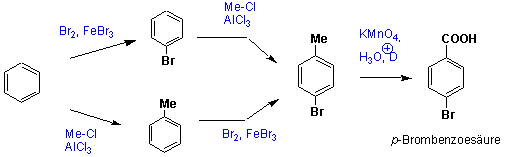
Aromatische Carbonsäuren
z.B. für die Synthese von para-Brombenzoesäure :
Aromatische Amine
Eine meta-dirigierende Nitrogruppe kann in die ortho- und para-dirigierende Aminogruppe umgewandelt werden. Nitrogruppen können zu Aminogruppen reduziert werden:
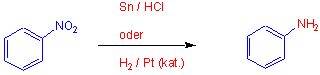
Für eine Synthese von p-Aminobenzoesäure (ein Bestandteil des Vitamins Tetrahydrofolsäure) :
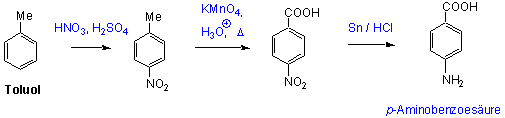
(Aufpassen: Friedel-Crafts Reaktionen an Nitrobenzol sind nicht möglich - -NO2 wirkt zu stark desaktivierend)
Durch Kondensation oder Annelierung mehrerer Benzolringe ergibt sich eine Verbindungsklasse, die man als mehrkernige benzoide Kohlenwasserstoffe bezeichnet.
z.B.
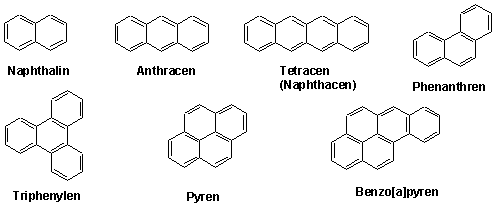
An diesen Ringen kann man, wie zu erwarten ist, elektrophile Substitutionen durchführen, die nach demselben Mechanismus wie die entsprechenden Reaktionen an Benzol und seinen Derivaten verlaufen. Die neue Hauptschwerpunkt liegt mit der regioselektivität solche Prozesse, die wir aber hier nicht betrachten werden.
Viele der mehrkernigen benzoiden Kohlenwasserstoffe sind karzinogen (krebserregend). Ein besonders gut erforschtes Molekül ist Benz[a]pyren. Benz[a]pyren entsteht bei der Verbrennung organischer Materie, wie Automobiltreibstoff und Erdöl, bei der Müllverbrennung, bei Waldbränden, man findet es in Zigaretten und im Zigarrenrauch und sogar in gegrilltem Fleisch.
Wodurch kommt nun die karzinogene Wirkung von Benz[a]pyren zustande ? Man nimmt an, dass ein oxidierendes Enzym (eine Oxidase) aus der Leber den Kohlenwasserstoff in das C7/C8 -Epoxid überführt. Ein anderes Enzym (Epoxid-Hydratase) katalysiert die Hydratisierung des Produkts zum trans-Diol (Vgl. Seite 35). Durch weitere Oxidation entsteht dann das eigentliche Karzinogen, ein neues C9/C10 -Epoxid:
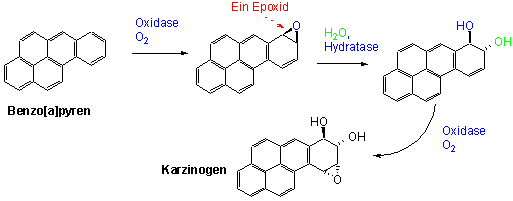
Vermutlich erfolgt das krebsauslösende Ereignis dann, wenn der Aminstickstoff des Guanins, einer der Basen im DNA-Strang, das Epoxid Nukleophil angreift :

Bei dieser Reaktion wird die Struktur eines Basenpaare der DNA geändert, was zu Fehlern und Störungen bei der DNA-Replikation führt. Diese Fehler können zu einer Veränderung (Mutation) des genetischen Information führen, wodurch dann unter Umstände das Wachstum einer Linie von rasch und undifferenziert wuchernden Zellen ausgelöst wird, was typisch für Krebs ist.
|

| Graphen ist die Bezeichnung für eine Modifikation des Kohlenstoffs mit zweidimensionaler Struktur, in der jedes Kohlenstoffatom von drei weiteren umgeben ist, so dass sich ein bienenwabenförmiges Muster ausbildet (siehe Bild links). Graphen hat ungewöhnliche Eigenschaften, die es sowohl für die Grundlagenforschung als auch für Anwendungen interessant machen, und zwar vor allem in der Physik (wie auch der Nobelpreis zeigt, der 2010 vergeben wurde). Beispielsweise sind Graphen-Flächeneinkristalle innerhalb der Flächen außerordentlich steif und fest. Wegen der hohen elektrischen Leitfähigkeit von Graphen wird derzeit an der Frage geforscht, ob Graphen Silicium als Transistormaterial ablösen könnte. |
