Research in the Hutter Group
Software Development: The CP2K Code
Simulation of Nanomaterials and Surfaces
The boron nitride nanomesh and related materials
For several years we have had a very successful collaboration with the group of Prof. Osterwalder and Prof. Greber at the Physics Department of the University of Zurich. Together, we investigate 2D materials obtained from the growth of graphene or hexagonal boron nitride (h-BN) at the surface of transition metals like Rh, Ru, Ni, and Cu. We addressed structural and electronic properties of the bare interfaces, the formation and dynamics of defects, intercalation processes, and adsorption of interesting molecules on such substrates (functionalisation).
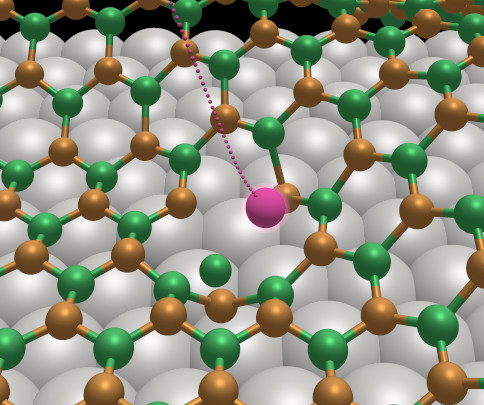
A large part of this collaboration has been focused on the so called h-BN/Rh nanomesh. The structure of hexagonal BN is closely related to that of graphite and is isoelectronic with the carbon polymorph. In 2003, Corso et al. [Science, 2004] discovered a new structure consisting of one single layer of h-BN adsorbed on Rh(111) surface. The nanomesh presents a highly regular, but corrugated, super-structure where hexagonal depressions (pores) with a diameter of about 2 nm are formed. In the pores, h-BN is bonded to the metallic surface, whereas around the pores the wires remain more distant from Rh and their interaction is dominated by dispersion contributions. Since 2008 we have been exploring various aspects of the h-BN nanomesh and related systems. In particular, we have used large-scale simuations in combination with experimental insights to address the following points:
- Construction and assessment of models for h-BN/Rh(111)
- Adsorption and confinement of water oligomers on h-BN/Rh(111)
- Study of the dehalogenation reaction of adsorbed molecules on h-BN/Rh(111)
- Simulating argon sputtering on the nanomesh and the resulting defect structures (figure)
- Wetting of liquid water on the nanomesh
Beyond h-BN/Rh heterostructures we have also investigated the electronic corrugation of h-BN/Cu(111), as well as adsorption and small-molecule activation on h-BN/Ni(111).
Marcella Iannuzzi, Ralph Koitz, Dorothea Golze
Two-dimensional metal-organic frameworks
Since the advent of graphene tremendous research efforts have focused on two-dimensional materials.
Such materials, made of a single molecular layer are of prime importance in nanotechnology, surface science and functional materials.
A promising new way to prepare such extended structures is through interlinking precursor molecules that have been adsorbed on a water surface.
Thus, functional and tunable 2D Metal-Organic Frameworks (MOFs) can be produced.
Our work focuses on studying the molecular details of the MOF formation on water using high-performance molecular dynamics simulations.
In particular, we are interested in the dynamic properties of monomers such as TTPB (a,b) on the surface of water, the process of ion migration from the liquid to the surface (c), and the energetics of interlinking TTPB into dimers and aggregates (d).
Ralph Koitz
Development of Advanced Electronic Structure Methods
MP2 and RPA methods for systems in the condensed phase
Currently Density Functional Theory (DFT) is the most widely used quantum mechanical tool for studying matter at the atomistic level.
However, approximate DFT still suffers from many flaws and the quest for finding more accurate functionals is still ongoing.
In particular, the inclusion of non-local dynamical electron correlation effects in the framework of DFT is one of the most active development lines in this field.
The second-order Møller-Plesset perturbation theory (MP2) and the Random Phase Approximation (RPA), are increasingly popular approaches in this context.
Both MP2 and RPA display many appealing features such as the capability to describe covalent, ionic, hydrogen-bond and dispersion interactions accurately and from first principles.
On the other hand, these advantages come at a computational cost that is significantly higher than traditional DFT.
On this basis, our research is focused on the development of efficient algorithms, capable to exploit the performance of the latest supercomputers,
in order to extend the range of applicability of these methods.
Additionally, this project includes benchmark and production calculations for which the novel approaches are used to study
complex and challenging systems such as molecular crystals, bulk liquid water and ice at high pressure.
Mauro Del Ben
Polarization and charge models for hybrid approaches
Hybrid quantum mechanics/molecular mechanics (QM/MM) description for molecules on surfaces are proposed where the molecules are treated at the QM and the substrate at the MM level of theory. If the substrate is metallic, induction effects are accounted for by applying the image charge formulation. The charges induced in the metal and the electrostatic response of the QM potential are determined self-consistently imposing the constant-potential condition in the metal. The image charge augmented QM/MM approach reproduces characteristic polarization effects of the adsorbates allowing to run large-scale simulations of liquid/metal interfaces at affordable computational cost.1
If the substrate is non-metallic and polar, the electrostatic properties of the surface are reproduced by assigning specifically generated partial charges to each substrate atom. The charges are obtained from restrained electrostatic potential (RESP) fits designed for periodic, slab-like systems. This approach was successfully employed to study the wetting of water on the h-BN/Rh(111) nanomesh.2
- D. Golze, M. Iannuzzi, M.-T. Nguyen, D. Passerone, J. Hutter, J. Chem. Theory Comput., 2013, 9, 5086-5097
- D. Golze, J. Hutter, M. Iannuzzi, Phys. Chem. Chem. Phys., DOI: 10.1039/c4cp04638b
Dorothea Golze
Local density fitting
A local density fitting technique is introduced for Kohn-Sham (KS) density functional calculations within a mixed Gaussian and plane waves approach (GPW). The atomic pair densities are approximated by an expansion in one-center fit functions reducing the scaling for the computation for two-electron integrals. The fitted density is employed for the calculation of Coulomb as well as exchange-correlation potential. Using local fitting within a GPW framework, improvements are obtained for the calculation of grid-dependent terms and the prefactor for building the KS matrix is reduced.
Dorothea Golze
Multi-objective parameter optimization techniques
The purpose of this research is to develop and test multi-objective optimization algorithms within the framework of quantum chemistry methods. Since parameter optimization is a ubiquitous topic in computational chemistry this affects all domains. Of particular interest are the semi-empirical methods given the implicit relevance of the parameter sets. By proposing a systematic approach we hope to improve the existing models and provide more transferable parameters.
Yannick Misteli
Simulation of Reactions and Catalysts for Sustainable Energy Production
TiO2-based Photo-catalyst Design for Water Reduction
Hydrogen production gains importance due to the world’s energy demand. It can be accomplished by photo-catalysis that converts energy of sunlight into H2 by reducing H2O. Among tested photo-catalysts for water reduction, TiO2 (Rutile phase) appears to be a promising one because of its simple preparation and stability. However, TiO2 has to be modified by adding photo-sensitizers and/or metal centers to decrease its large band gap.
We aim to theoretically design an efficient TiO2-based photo-catalyst for hydrogen production by water reduction. To modify the band gap of TiO2 and increase its photo-catalytic activity, a pyridine-based molecule is used as photo-sensitizer and a cobalt atom as metal center. Rutile has three major crystallographic faces, which are (110), (101), and (100) with a ratio of 60 %, 20 %, and 20 %, respectively. Among these major faces, (110) has the lowest surface energy, therefore surface calculations are carried out using Rutile(110). Our strategy for the investigation of adsorption on TiO2 surface is first optimizing the structure at the PBE level of theory, afterwards obtaining more accurate electronic structure of the optimized systems with the Heyd-Scuseria-Ernzerhof (HSE06) hybrid functional.
Yeliz Gürdal
Exploring Co(II)-based Cubanes for Water Oxidation
We investigate Co(II)-based cubanes, which are employed as water oxidation catalysts (WOCs) in artificial photosynthesis, by computational methods encompassing static and dynamic approaches.
As a first step, we investigated energy differences, reaction barriers, free energy changes, and possible reaction pathways for ligand exchange reactions of Co(II)-based cubanes where acetate ligands are replaced by either water or a hydroxide. With this work, we aimed at the elucidation of the exact ground state structure for the catalytic cycle. We studied the stability of many different configurations through comparison of energies obtained from Born-Oppenheimer Molecular Dynamics (BOMD) sampling of the respective molecules in a periodic box of water and also used methods such as the nudged elastic band and metadynamics.
We further focus on a critical assessment of the effects of solvation on the energetics of water oxidation reactions as well as a reliable computation of redox reactions occurring during the catalytic cycle.
The general goal of our work is to understand the details and energetics of complete reaction mechanisms, determine the most important factors influencing WOC activity and, with this knowledge at hand, to finally design efficient and robust WOCs.
Florian Hodel
High-Performance Ab-initio Molecular Dynamics
Vibrational Spectroscopy and Molecular Dynamics
In the context of spectroscopy we are particularly interested in vibrational spectroscopy. Here, we mainly rely on density functional theory-based molecular dynamics and focus on periodic systems such as liquids. Recently, the CP2K program has been extended to efficiently calculate Raman property tensors as well as their decomposition into intra- and intermolecular contributions. As an example, the picture shows the computed Raman spectra of liquid (S)-methyloxirane (for details, see this paper).
Sandra Luber