 Inhalt Index
Inhalt IndexDie Kenntnis der zahlreichen organischen Reaktionen und funktionellen Gruppen, die in den vorangegangenen Kapiteln vorgestellt wurden, sollten Sie befähigen manches komplizierte Molekül im Laboratorium herzustellen. Wie können wir jedoch sicherstellen, dass es sich bei der dargestellten Verbindung tatsächlich um die gewünschte Substanz handelt ? Dieselbe Frage entsteht, wenn eine organische Verbindung aus biologischem Material erhalten worden ist (was ist seine Struktur?). Zur Beantwortung dieser Fragen stehen eine Reihe von Messmethoden und chemischen Nachweisen zur Verfügung.
z.B. physikalische Eigenschaften als Beweis für Reinheit :
Verhalten bei der Chromatographie (Säulenchromatographie, Papierchromatographie, Dünnschichtchromatographie, Gaschromatographie, Hochdruck-Flüssigkeits-Chromatographie (HPLC), Gelchromatographie, Destillation (Siedepunkt), Kristallisation (Schmelzpunkt), Optisches Drehvermögen (wenn chiral).
Erst wenn eine organische Verbindung gereinigt und durch ihre physikalische Eigenschaften als reiner Stoff charakterisiert worden ist, kann ihre chemische Zusammensetzung ermittelt werden. Dazu ist es notwendig eine quantitative Elementaranalyse durchzuführen, d.h. den prozentualen Anteil jedes der in der betreffenden Substanz enthaltenen Elemente zu bestimmen. Zur Bestimmung von C, H und N bedient man sich auch heute noch der Verbrennungsanalyse. Die Substanz wird in einem Verbrennungsofen verbrannt, wodurch durch verschiedene Methoden C, H und N, in CO2, H2O oder N2 übergeführt werden. Die Verbrennungsprodukte werden in Absorptionsröhrchen aufgefangen und gewogen. Der Sauerstoffgehalt einer Verbindung wird meistens indirekt bestimmt (der fehlende Rest !). Aus der prozentualen Zusammensetzung einer Substanz lässt sich ihre empirische Formel berechnen, z.B:
Eine aus C, H und O bestehende Substanz besitzt folgende Zusammensetzung:
C: 61.45% H: 7.72% O: 30.83%
Die Koeffizienten der einzelnen Symbole in der Substanzformel erhält man, indem man den prozentualen Anteil durch die jeweilige Molmasse dividiert:
C: 61.45/12 = 5.12 H: 7.72/1 = 7.72 O: 30.83/16 = 1.92
Substanzformel: C5.12H7.72O1.92
Division dieser Koeffizienten durch die kleinste Zahl ergibt: C2.66H4.02O
Durch Multiplikation mit der kleinstmöglichen ganzen Zahl werden die gebrochenen Koeffizienten möglichst annähernd in ganze Zahlen umgewandelt:
3x = C7.98H12.06O3 d.h. C8H12O3
Die geringen Abweichungen der berechneten Koeffizienten von ganzzahligen Werten widerspiegeln den experimentellen Fehler (ca. 0.3%). Seine Grösse wird durch einen Vergleich der berechneten und der gemessenen prozentualen Zusammensetzung deutlich :
berechnet für C8H12O3 C: 61.53% gefunden C: 61.45%
H: 7.69% H: 7.72% O: 30.78% O: 30.83%
Was können wir tun, wenn wir zum Beispiel aus einer Reaktion (oder aus einem Lebewesen) eine Substanz der Summelformel C7H16O erhalten. Durch chemische Teste wäre es durchaus möglich zu schliessen, ob z.B. diese Verbindung ein Alkohol ist. Aber immerhin wüssten wir noch wenig über die Struktur der Verbindung.
In der modernen Organischen Chemie (und Molekularbiologie) steht ein weiteres Mittel zur Lösung solcher Probleme zur Verfügung : Die Spektroskopie.
Es gibt verschiedene Arten von Spektroskopie, von denen vier in der organischen Chemie besonders wichtig sind:
Kernresonanz-Spektroskopie (NMR : nuclear magnetic resonance)
Infrarot-Spektroskopie (IR)
Ultraviolett-Spektroskopie (UV)
Massenspektroskopie (MS)
Organische Moleküle absorbieren Strahlung in diskreten "Portionen"DE = h n, die auch Quanten genannt werden. Die absorbierte Strahlung bewirkt im Molekül eine Veränderung des elektronischen oder mechanischen Zustandes, ein Prozess, der auch als Anregung bezeichnet wird :
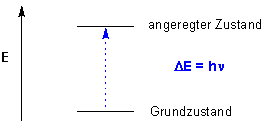
Die unterschiedlichen Strahlungsarten, die damit verbundenen Energiebeträge und die dementsprechenden Wellenlängen sind unten zusammengestellt:
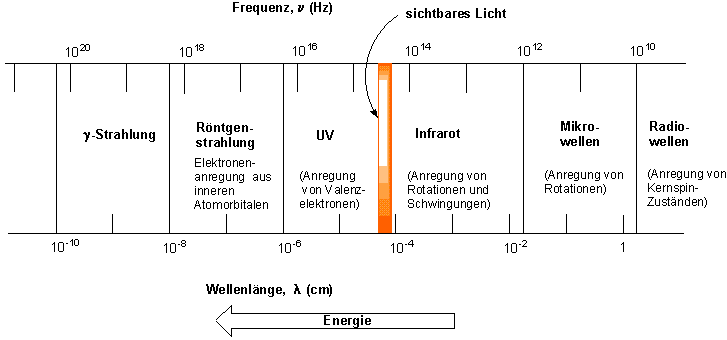
Man erinnere sich, dass die Energie von Strahlung mit der Frequenz n and der Wellenzahl (= 1/l) ansteigt, mit steigender Wellenlänge l jedoch abnimmt. Ein Molekül kann vielen unterschiedlichen Arten der Anregung unterliegen, von denen eine jede ihre genau bestimmte Energie DE erfordert. Infrarot-Strahlung bewirkt die Anregung von Schwingungen im Molekülgerüst einer Verbindung. Radiowellen sind in der Lage, die Orientierung von Kernspins in einem Magnetfeld umzukehren. Dieses Phänomen stellt die Grundlage der NMR-Spektroskopie dar.
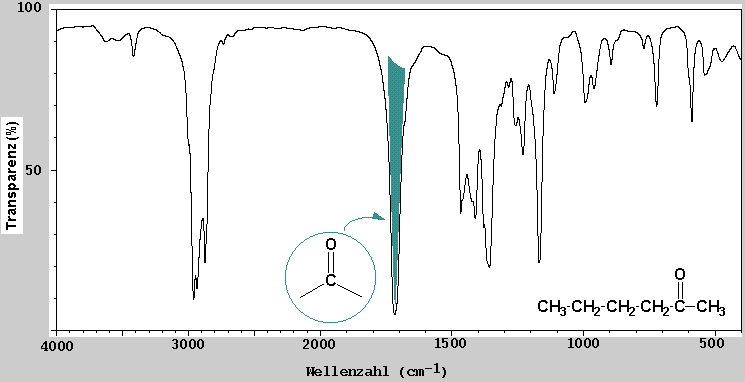
Der IR-Bereich des elektromagnetischen Spektrums von 2.5 x 10-3 (= 2.5 �m = 4000 cm-1) bis 2.5 x 10-4 cm (=25 �m = 400cm-1) wird normalerweise für IR-Spektroskopie benutzt. Die Anregung von Molekülschwingungen kann man sich anhand eines einfachen Modells veranschaulichen. Die Atome A und B seien wie zwei Massen an einer Feder miteinander verbunden. Diese Feder dehnt sich und zieht sich mit einer bestimmten Frequenz n zusammen. In diesem Bild hängt die Frequenz der beiden Atomschwingungen, von der Stärke der Bindung zwischen ihnen und ihrer Atommasse ab. Tatsächlich lässt sich diese Bewegung mit Hilfe des Hookschen Gesetzes beschreiben :
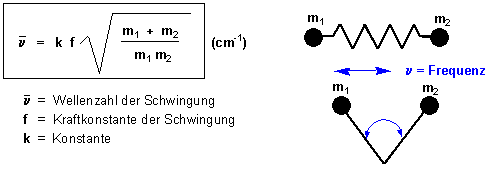
IR-Spektren sind jedoch komplexer als zu erwarten wäre, wenn man nur die Anzahl von unterschiedlichen Bindungen eines Moleküls zählt. Der Hauptgrund für die Komplexität liegt in der Vielzahl von möglichen Schwingungen und in der Tatsache, dass viele Schwingungen mechanisch gekopplt sind. Ein IR-Spektrum enthält normalerweise viele Peaks und eine Interpretation des gesamten Spektrums ist deswegen recht schwierig. Dennoch ist die IR-Spektroskopie in der Praxis aus zwei Gründen von grossen Nutzen:
- erstens erscheinen die Schwingungsbänder einer Reihe von funktionellen Gruppen bei charakteristischer Wellenzahl
- zweitens lässt sich das gesamte Spektrum zur Identifizierung einer Verbindung verwenden, da jede Substanz ihr eigenes, unverwechselbares Spektrum hat.
Interaktive Visualisierung von Valenzschwingungen organischer Moleküle
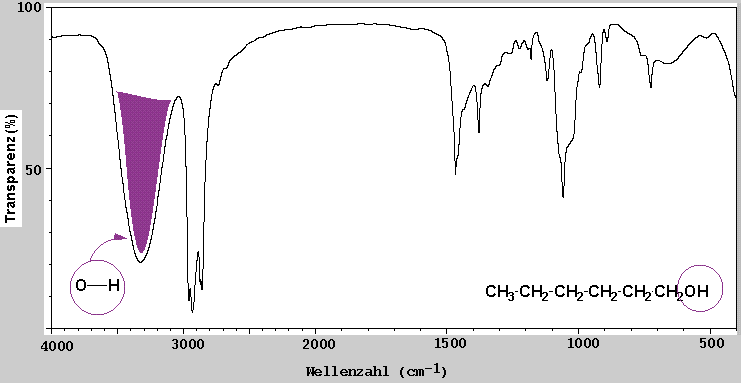
Wenn z.B. eine Substanz keinen Peak bei 3600-3200 cm-1 in dem IR-Spektrum zeigt, kann man daraus schliessen dass keine O-H Gruppen vorhanden ist.
Bei Verbindungen die eine Keton-Carbonylgruppe enthalten, erscheint analogerweise immer ein Peak im Bereich 1725-1710 cm-1. Es ist deshalb notwendig zu wissen, bei welchem Wellenzahlbereich die charakteristischen Valenzschwingungen der üblichen funktionellen Gruppen in einem IR-Spektrum erscheinen.
Wellenzahlbereich von charakteristischen Valenzschwingungen organischer Moleküle
| Funktionelle Gruppe | Wellenzahl cm-1 | Intensität | Valenz |
|---|---|---|---|
| Alkane, Alkylgruppen | 2850-2960 | stark | } C-H |
| Alkene | 3020-3100 | mittel | |
| 1650-1670 | mittel | C=C | |
| Alkine | 3300 | stark | 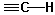
|
| 2100-2260 | mittel | 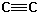
| |
| Alkylhalogene | 600-800 | stark | C-Cl |
| 500-600 | stark | C-Br | |
| 500 | stark | C-I | |
| Alkohole | 3200-3600 | stark, breit | O-H |
| 1050-1150 | stark | C-O | |
| Aromaten | 3030 | mittel | C-H |
| 1600,1500 | stark | 
| |
| Amine | 3310-3500 | mittel | N-H |
| 1030,1230 | mittel | C-N | |
| Carbonyl-Verbindungen | 1670-1800 | stark | C=O |
| Carbonsäuren | 2500-3100 | stark, sehr breit | O-H |
| Nitril | 2210-2260 | mittel | 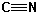
|
| Nitrogruppen | 1560,1350 | stark | -NO2 |
Es ist manchmal hilfreich die IR-Absorptionen in vier Bereiche zu unterteilen :
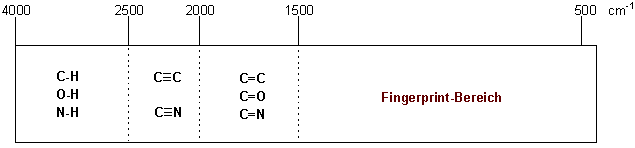
Im Bereich von unter 1500 cm-1, dem sogenannten Fingerprint-Bereich, findet eine Vielzahl von Absorptionen verschiedener C-O, C-C und C-N Schwingungen statt. Diese Peaks bilden ein Muster, das für eine bestimmte Verbindung charakteristisch ist, einen Fingerabdruck ("Fingerprint").
Die Ultraviolett-Spektroskopie, die im Bereich von 200-400 nm arbeitet, und die Spektroskopie im sichtbaren Bereich (Vis-Spektroskopie), die die Strahlung von 400-800nm verwendet, sind für die Untersuchung der elektronischen Struktur von ungesättigten Molekülen und zur Messung des Ausmasses ihrer Konjugation wichtig. So erhaltene Spektren nennt man Elektronenspektren.
Der Vorgang, der durch eine derartige Absorption von Strahlung ausgelöst wird, ist die Anregung von Elektronen aus besetzten bindenden und nichtbindenden Molekülorbitalen in unbesetzte antibindende Molekülorbitale:
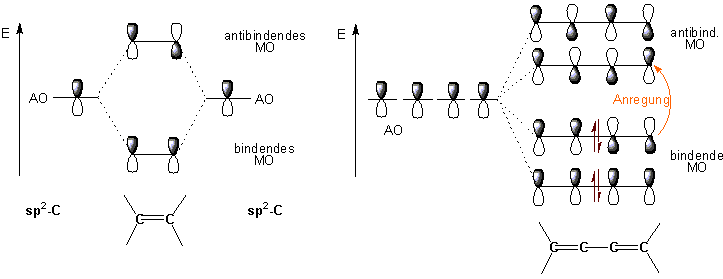
Die Anregung eines Elektrons in eine normale s-Bindung heisst s --> s* Übergang, wobei die Wellenlänge der absorbierten Strahlung üblicherweise im extremen bzw, Vakuum -UV-Bereich unter 200 nm liegt (spezielle Ausstattung nötig). Im Gegensatz dazu erfordert die Anregung der Elektronen in p-Bindungen weniger Energie und kann im Bereich über 200 nm beobachtet werden (p --> p* Übergängen). Auch nichtbindende Elektronen können in diesem Bereich in höhere Niveaus angeregt werden (n -->s* oder n --> p* Übergängen).
z.B.
| C-C, C-H | s --> s* | ~ 150 nm |
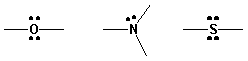 | n --> s* | ~ 185 nm |
 | n --> s* | ~ 195 nm |
| n --> p* | ~ 300 nm | |
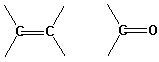 | p --> p* | ~ 190 nm |
Ein konjugiertes Molekül kann eine Vielzahl von bindenden, nicht-bindenden und antibindenden Molekülorbitalen haben. Daher sind zahlreiche Übergänge möglich. Man findet die entsprechenden Absorptionssignale für energiereiche Übergänge im Bereich der kürzeren Wellenlängen des Spektrums, die Signale für weniger energieaufwendige Übergänge erscheinen bei grösseren Wellenlängen.
Ein Signal im UV-Spektrum wird durch die Lage des Absorptionsmaximums (der Bandenspitze) lmax in nm charakterisiert. Die Höhe des Signals ist die Extinktion E (oder A = Absorption). Sie wird in Form des für das Molekül charakteristischen Extinktionskoefficienten e angegeben :

Konjugierte Systeme, die bei Wellenlängen über 400 nm absorbieren, sind farbig. z.B. sind Moleküle, die bei 450 nm absorbieren orangerot, jene die bei 550 nm absorbieren violett, und jene die bei 650 nm absorbieren blaugrün.
Elektronenspektren können zeigen, inwieweit ein Molekül konjugiert ist. Je mehr Doppelbindungen in Konjugation stehen, desto grösser wird die Wellenlänge für die energieärmste Absorption sein, und desto mehr Signale werden im Spektrum auftreten, z.B.:
| n | lmax | e | |
|---|---|---|---|
| 3 | 275 | 30,000 | Me-[-CH=CH-]n-Me vgl. UV-Spektren |
| 4 | 310 | 76,000 | |
| 5 | 342 | 122,000 | |
| 6 | 380 | 146,500 |
Warum hat die Zahl der konjugierten Doppelbindungen einen derart starken Einfluss auf das Elektronenspektrum ? Mit ansteigendem Grad der Konjugation wächst die Zahl der Energieniveaus, die zu den entsprechenden p-Orbitalen gehören. Entsprechend sinkt der Energieunterschied zwischen dem höchsten besetzten und dem niedrigsten unbesetzten MO. Ausgedehntere p-Systeme erfordern daher zur elektronischen Anregung weniger energiereiche Strahlung von grösserer Wellenlänge.
Mit zunehmender Konjugation und zunehmender Substitution wächst die Wellenlänge des Absorptionsmaximums lmax. Sein Wert ist auch für bestimmte Strukturtypen charakteristisch.
Eine interessante Anwendung von UV-absorbierenden Molekülen liegt bei der Herstellung von Sonnenschutzmitteln. Der UV-Vis Bereich des Sonnenlichts wird in verschiedenen Regionen klassifiziert :

Chromophore, die für die Absorption von UV-A oder UV-B Strahlen geeiget sind und kommerziell hergestellt werden, sind unter anderem:
| UV-B | UV-A |
|---|---|
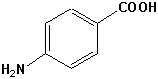 p-Aminobenzoesäure | 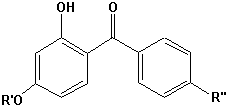 Benzophenone |
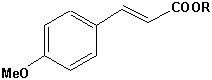 Zimtsäureester | 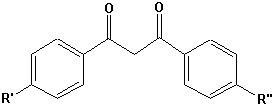 Dibenzoylmethan |
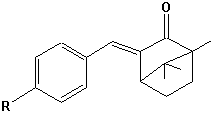 Benzylidencampher | 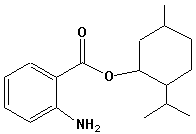 Menthyl-Anthranilsäureester |
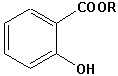 Salicylsäureester |
Die entsprechenden UV-Spektren von drei solchen Verbindungen sind auf einem separaten Blatt wiedergegeben.
Die für die Strukturaufklärung wichtigste Methode ist die NMR-Spektroskopie.
18.3.1 Kernspins und die Absorption von Radiowellen
Viele Atomkerne (am wichtigsten 1H und 13C) verhalten sich so, als würden sie sich um ihre eigene Achse drehen. Man sagt daher, dass sie einen Kernspin haben. Der Atomkern von 1H ist positiv geladen. Seine Rotation verursacht ein magnetisches Moment. Das bedeutet vereinfacht, dass man das Proton als einen kleinen Stabmagneten betrachten kann :
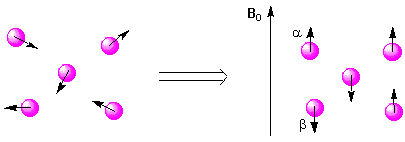
Setzt man das Proton einem äusseren Magnetfeld B0 aus, kann es im Prinzip eine von zwei Orientierungen einnehmen. Die energiearme Orientierung ist die zum Magnetfeld parallele, während die zum Magnetfeld antiparallele energetisch weniger vorteilhaft ist. Der Übergang erfordert die Zufuhr eines entsprechenden Energiequantums :

Um den Übergang zu messen, bedarf es einer Strahlung geeigneter Frequenz. Dann kommt es zur Resonanz, d.h. die zum Übergang von Spin a nach Spin b erforderliche Energie wird von der Probe aufgenommen, was sich spektroskopisch als Energieabsorption beobachten lässt.
Die Frequenz der absorbierten Strahlung n ist der Stärke des äusseren Magnetfelds B0 mit einer Konstante g direkt proportional :
n = g B0
g = Gyromagnetisches Verhältnis
Dabei ist die Konstante g für die Art des betrachteten Atomkerns und seine Umgebung charakteristisch. Die heute käuflichen Magneten weisen Feldstärken zwischen 1.4 und 15 Tesla (T) auf. Die entsprechenden Frequenzen n (für Protonen !) reichen von 60 bis 600 MHz.
Der Zusammenhang zur absorbierten Energie ergibt sich aus der Gleichung :
DEa-b = h n ~ 3.8 x 10-5 kJ/mol
d.h. DE ist von einem sehr sehr kleinen Wert.
Wenn wir 1H oder 13C NMR-Spektren betrachten, kann man sofort sehen, dass für die verschiedenen Wasserstoffatome oder 13C-Atome in einer Verbindung unterschiedliche NMR-Signale beobachtet werden. Warum ?
z.B.: CH3COOCH3
a) 1H NMR-Spektrum
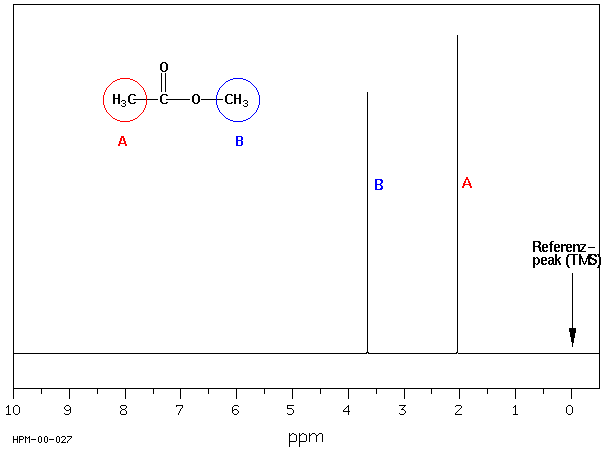
b) 13C NMR-Spektrum
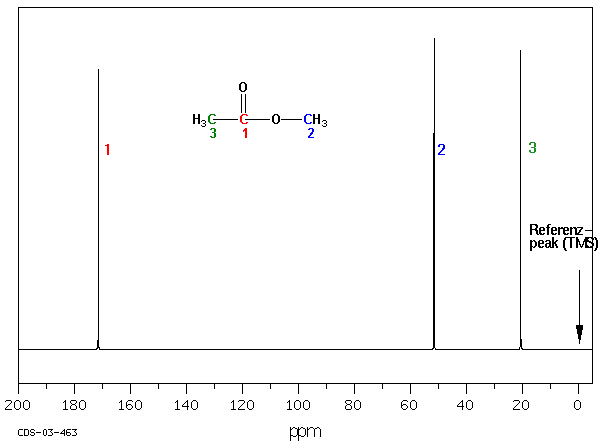
Die Ursache liegt in der unterschiedlichen elektronischen Umgebung der jeweiligen 1H- oder 13C-Atome. Die Kerne chemisch gebundener H-Atome sind von Elektronenwolken umgeben, deren Elektronendichte je nach Polarität der Bindung, der Hybridisierung der benachbarten Atome und der An- oder Abwesenheit elektronenziehender oder -liefernder Gruppen variiert. Wenn ein von Elektronen umgebener Atomkern einem Magnetfeld ausgesetzt wird, induzieren diese ein lokales Magnetfeld blokal, welches dem äusseren Magnetfeld B0 entgegen gerichtet ist :
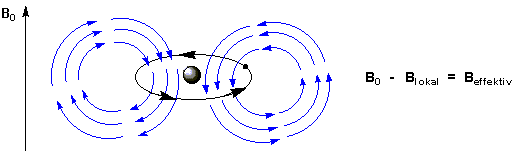
Die Konsequenz ist eine geringere effektive Feldstärke in der Umgebung des Wasserstoff-Atomkerns, der so vom äusseren Feld B0 durch die Elektronenwolke abgeschirmt wird.
Wenn das Spektrum bei konstanter Frequenz und variabler Feldstärke aufgenommen wird, stellt man fest, dass zur Kernresonance eine höhere Feldstärke erforderlich ist, um die Abschirmung zu überwinden (oder bei konstanter Feldstärke, eine Erniedrigung der Frequenz). Eine Verschiebung eines Signals nach höherem Feld bedeutet also, dass das Signal im Spektrum nach rechts verschoben wird :
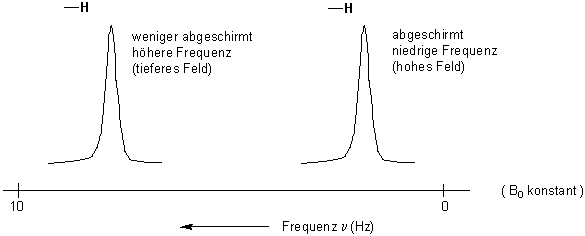
Ein jedes H-Atom (oder 13C-Atom für 13C-NMR Spektren) eines Moleküls hat entsprechend seiner elektronischen Umgebung eine bestimmte Resonanzfrequenz. Chemisch äquivalente H-Atome führen zu gleichen Signalen.
Anstatt nun die exakte Frequenz eines jeden Signals zu vermessen, wird meist eine kleine Menge eines internen Standards zur Probe gegeben. Die Lage der NMR-Signale der untersuchenden Substanz wird dann relativ zu den Signalen des internen Standards angegeben. Als interner Standard dient Tetramethylsilan Me4Si (TMS).
Dabei hat man allerdings das Problem, dass die Frequenzen je nach der Stärke des angelegten Magnetfeldes variieren, da Feldstärke und Resonanzfrequenz zueinander proportional sind. Um diese Komplikation zu umgehen, bildet man den Quotienten aus dem Frequenzunterschied zum Signal des TMS und der Messfrequenz des jeweiligen Spektrometers :
| d = | Frequenzunterschied zum Signal vom TMS (Hz) ____________________________________ Spektrometerfrequenz (MHz) | ppm |
Dies führt zu einer von der Feldstärke unabhängigen Grösse, der chemischen Verschiebung d.
Z.B.:
Für den internen Standard TMS ist d per Definition 0.00. Dieses System hat den enormen Vorteil, dass bei unterschiedlichen Feldstärken gemessene Spektren direkt miteinander vergleichbar sind. Ein Proton das d = 2.0 in einem 60 MHz Spektrum erscheint (2ppm x 60 MHz = 120 Hz von TMS), erscheint auch bei d = 2.0 in einem 500 MHz Spektrum, obwohl die Resonanzfrequenz jetzt 2ppm x 500 MHz = 1000 Hz von TMS entfernt liegt.
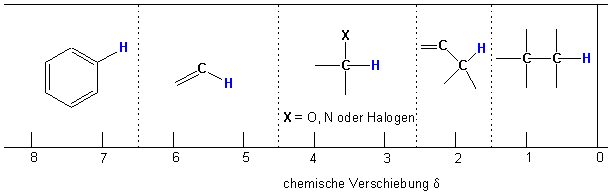
Der grosse Wert der NMR-Spektroskopie liegt darin, dass die chemische Verschiebung für die chemische (strukturelle) Umgebung der betreffenden Kerne charakteristisch ist .
Man beachte, dass die Signale der Alkane bei ziemlich hohem Feld erscheinen (d = 0.8 - 1.7 ppm). Zunehmende Substitution oder die Nähe eines elektronegativen Atoms - eine elektronenärmere Umgebung also - führt zu einer Absorption bei tieferem Feld (grösseren ppm-Werten). Eine elektronenreiche Umgebung verursacht den umgekehrten Effekt.
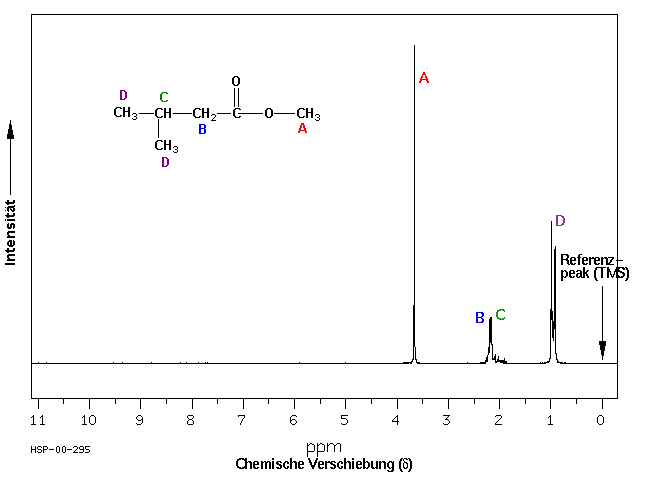
18.3.3 Die Integration als Hilfsmittel
Der Vergleich der Signalintensitäten in den NMR Spektren zeigt, dass die relative Signalintensität mit der Anzahl der dem Signal entsprechenden Kerne korreliert. Je mehr Wasserstoffatome einer Art in einem Molekül vorhanden sind, desto intensiver ist das dazugehörige NMR-Signal relativ zu den Signalen anderer Wasserstoffatome.
Die Zuordnung von Signalen aufgrund ihrer chemischen Verschiebung ist in Verbindung mit der Integration ein ausserordentlich wertvolles Hilfsmittel bei der Strukturaufklärung.
Die bisher behandelten Spektren hatten einfache Linienmuster : scharfe Singuletts. Aber wenn zwei nichtäquivalente Kerne in direkter Nachbarschaft zueinander stehen, führt das zu komplizierteren Spektren, da es dann zur Spin-Spin-Kopplung oder Spin-Spin-Aufspaltung kommt.
z.B.
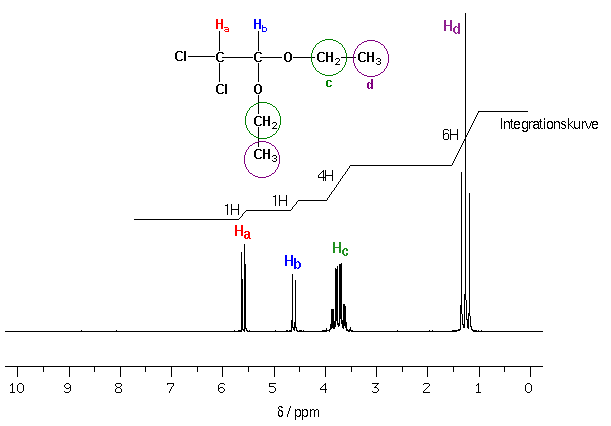
Wenn ein H-Atom Ha ein Hb zum Nachbarn hat, welches in Feldrichtung ausgerichtet ist, so ist Ha nicht nur dem externen Magnetfeld B0 ausgesetzt, sondern erfährt durch den a-Spin von Hb eine kleine Verstärkung des effektiven Magnetfelds. Man beobachtet dann, wie erwartet das entsprechende NMR-Signal bei tieferem Feld:
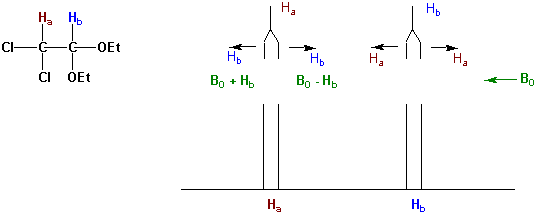
Aber die Hälfte der in der Probe enthaltenen Ha-Atome haben Atome-Hb mit b-Spin zum Nachbarn, also mit einer Orientierung gegen die Richtung des äusseren Feldes. Das lokale Feld bei Ha wird in diesem Fall geschwächt, und wie erwartet, beobachtet man im Spektrum eine Verschiebung nach höherem Feld.
Man sagt, das Signal eines Ha sei durch die Nachbaratome in ein Dublett "aufgespalten".
Entsprechende Überlegungen gelten für andere Situationen, zum Beispiel die O-CH2-CH3 Gruppe :
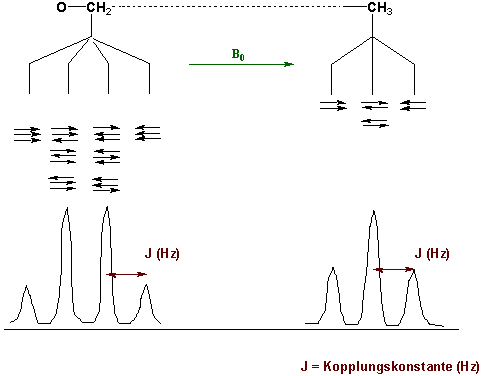
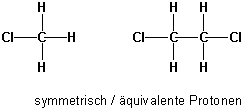
Man beachte, dass bei Kernen mit identischer chemischer Verschiebung (chemisch äquivalente Protonen) keine Spin-Spin-Kopplungen beobachtet werden können :
Die Überlegungen zur Spin-Spin-Aufspaltung führen zu einfachen Regeln : Kerne mit N-äquivalenten Nachbarn zeigen eine Aufspaltung in N+1 Linien. Ihre relativen Intensitäten lassen sich anhand des Pascalschen Dreiecks leicht berechnen:
| s (singulett) d (Dublett) t (Triplett) q (Quartett) quin (Quintett) u.s.w. | 1 1 : 1 1 : 2 : 1 1 : 3 : 3 : 1 1 : 4 : 6 : 4 : 1 u.s.w. |
Es ist wichtig sich daran zu erinnern, dass nicht äquivalente Kerne miteinander koppeln und ihre Signale gegenseitig aufgespalten sind. Der Abstand der Linien in einem Aufspaltungsmuster ist die Kopplungskonstante, J in Hz. Für zwei miteinander koppelnden Kerne sind die Kopplungskonstanten für beide Signale gleich:
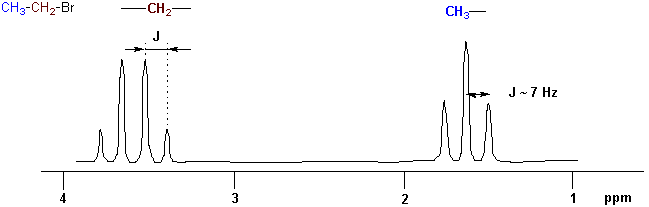
Die Kopplungskonstante ist unabhängig von der Stärke des externen Magnetfeldes.
Weitere Beispiele : 1-Nitropropan
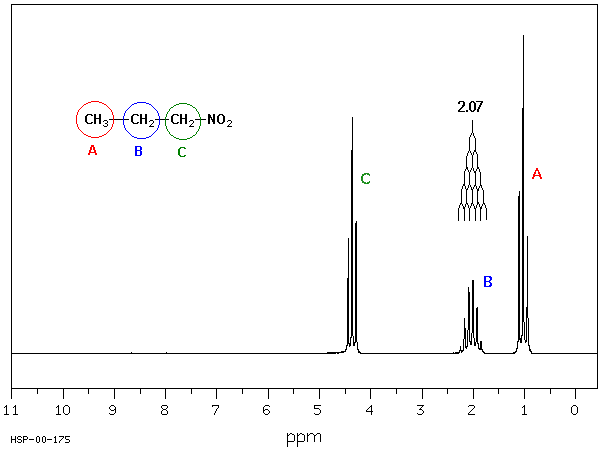
Das häufigste Isotop des Kohlenstoffs, 12C, ist NMR-spektroskopisch nicht nachweisbar. Aber das 13C-Isotop tritt mit einer Häufigkeit von 1.11% auf, und im Magnetfeld verhält es sich ähnlich dem Isotop 1H. Es ist wegen der erheblich geringeren natürlichen Häufigkeit des Isotops 13C und der geringeren Empfindlichkeit von 13C schwieriger 13C-NMR-Spektren aufzunehmen.
Ein Vorteil der geringen natürlichen Häufigkeit des Isotops 13C liegt darin, dass in normalen 13C NMR-Spektren keine C-C-Kopplungen auftreten. Im einfachsten Fall liefert jedes nicht-äquivalente C-Atom in einem Molekül eine Linie im 13C-NMR-Spektrum :
Z.B.: 4'-Bromacetophenon
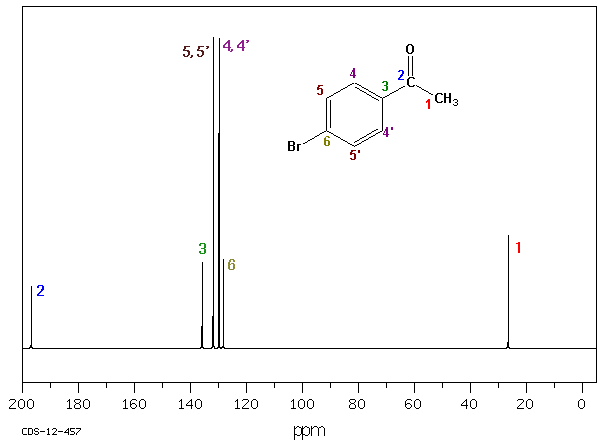
In diesem Fall zeigt das 13C-NMR-Spektrum nur 6 Linien, die den 6 magnetisch nichtäquivalenten C-Atomen des Moleküls entsprechen.
Ähnlich der 1H NMR-Spektroskopie, werden die chemischen Verschiebungen in der 13C-NMR-Spektroskopie durch die chemische Umgebung des jeweiligen Kohlenstoffatoms bestimmt. Der Bereich der chemischen Verschiebungen ist jedoch mit etwa 200 ppm deutlich grösser als in der 1H-NMR-Spektroskopie.
