Atomistic description of reactive processes in heterogeneous catalysis
.png)
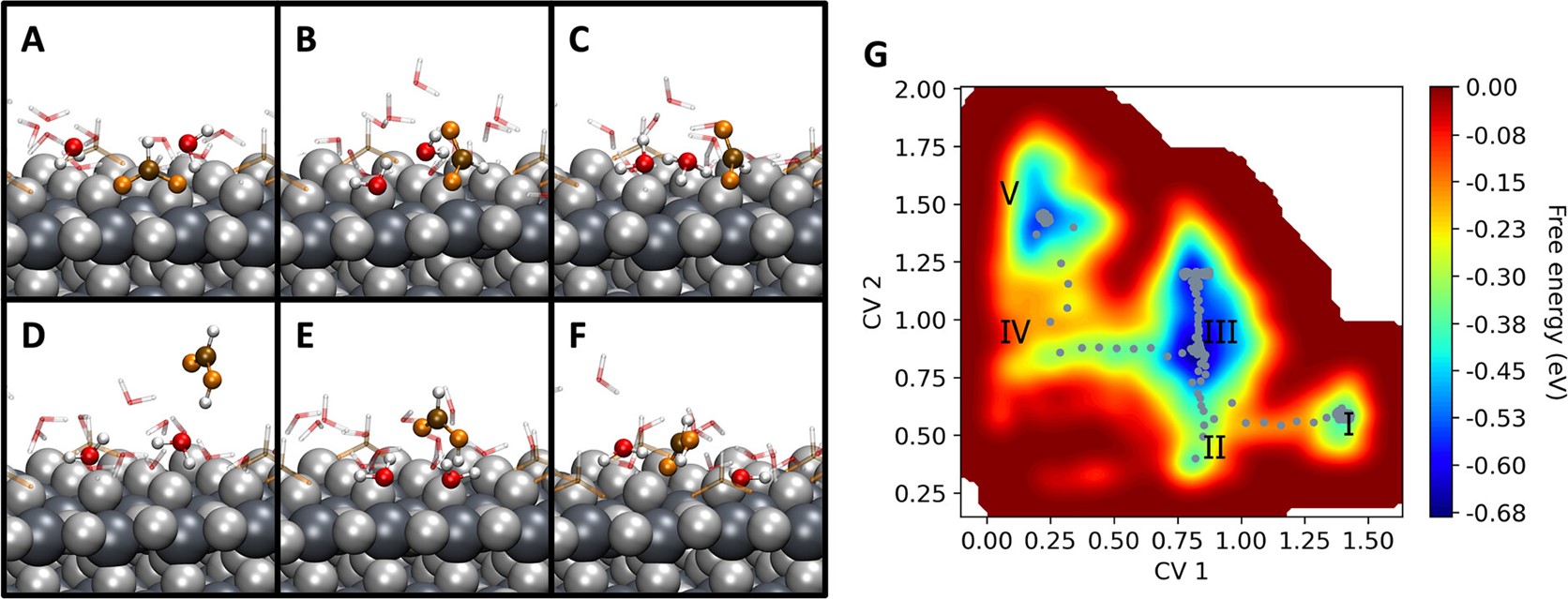
To date, the interactions of gas/solid, gas/solid/solid, and solid/liquid interfaces under realistic working conditions (temperature, pressure, potential) have been studied extensively using classical potentials, making electronic-level insight impossible. Only recently has it become feasible to use electronic structure theory, mainly DFT, for such extensive systems in surface science. This has enabled a deeper understanding of the dynamics at interfaces, by treating interactions on a balanced quantum mechanical footing whilst achieving the necessary statistical sampling to evaluate free energies and describe processes. Nevertheless, the atomistic simulation approach has still to fill an important gap. Under working conditions, adsorbates are mobile and complex adsorbate−adsorbate interactions depending on the distribution of surface species take place, which can significantly impact the energetics of surface reactions and their most favored pathways. Common strategies are by refining the available experimental information by comparing computed and measured spectroscopic signatures and thermodynamic approaches, where the stability of candidates under given conditions is assessed via ab initio thermodynamics. Combining the accurate description of the interatomic interactions, including environment effects (explicit solvent and electrified interfaces), with the sampling of the accessible phase space, using ab initio-level molecular dynamics and metadynamics, we shed light on the morphological changes of active sites, adsorption, selective co-adsorption and overall reactive processes at the interface. We aim at the simulation of complete experimental setups, i.e., support material coupled with reactive species and in contact with a solution. A complete model of this type will have a size that is close to the limit of what is currently possible. It is therefore necessary to carefully build up models that, without compromising on accuracy, allow for extended simulations. Employing characterization tools based on theoretical spectroscopy (XAS, XPS, IR/Raman) for the analysis of trajectories and specific structures, we will gain insight into the structure/property relation and improve understanding and design of experiments. We will address the dynamics and stability of the intermediates of catalytic processes, with a focus on electron transfer reactions across interfaces which is of high relevance in electrochemistry and catalysis.