Elucidating the solution structure of the monomolecular BCL2 RNA G-quadruplex: a new robust NMR assignment approach Chem. Sci. , 2025.
DOI: 10.1039/D5SC01416F
5′ untranslated regions (UTRs) of mRNA commonly feature G-quadruplexes (G4s), crucial for translational regulation and promising as drug targets to modulate gene expression. While NMR spectroscopy is well-suited for studying these motifs' structure and dynamics, their guanine-rich nature complicates resonance assignment due to high signal overlap. Exploiting the inherent rigidity of G4 cores, we developed a universally applicable assignment strategy for uniformly isotopically enriched G4 structures, relying solely on through-bond correlations to establish the G-tetrads. Applying this approach, we resolved the solution structures of two triple mutants of the RNA G4 in the 5′ UTR of the human BCL2 proto-oncogene, one of the first natural monomolecular RNA G4 structures available to date. Comparative analysis with other RNA and DNA G4s reveals their notably compact and well-defined cores. Moreover, the sugar pucker geometries of the tetrad guanines are far less stringent than previously assumed, adeptly accommodating specific structural features. This contrasts with the canonical base pairing in RNA and DNA, in which the sugar pucker dictates the type of the double-helical structure. The strategy presented provides a direct path to uncovering G4 structural intricacies, advancing our grasp of their biological roles, and paving the way for RNA-targeted therapeutics.
2024
Fluorescent End-Labeling and Encapsulation of Long RNAs for Single-Molecule FRET-TIRF Microscopy
Ahunbay, Esra; Fazliji, Besim; Sivanantharasa, Nirusan; Sigel, Roland K. O., J. Vis. Exp. 2024, 212 , e67391.
Single-molecule Förster Resonance Energy Transfer (smFRET) excels in studying dynamic biomolecules by allowing precise observation of their conformational changes over time. To monitor RNA dynamics with smFRET, we developed a method to covalently label RNAs at their termini with a FRET pair of fluorophores. This direct end-labeling strategy targets the 5'-phosphate by carbodiimide (EDC)/N-hydroxysuccinimide (NHS) activation and the 3'-ribose by periodate oxidation, which can be adapted to other RNAs regardless of their size and sequence to study them independently of artificial modifications. Furthermore, the 5'-EDC/NHS activation is of general interest to all nucleic acids with a 5'-phosphate. The use of commercially available chemicals eliminates the need to synthesize RNA-specific probes. Total Internal Reflection Fluorescence (TIRF) microscopy requires the surface-immobilized molecules of interest to be within the evanescent field to be illuminated. A sophisticated way of keeping the RNA molecules within the evanescent field is to encapsulate them in phospholipid vesicles. Encapsulation benefits from the best of both worlds, tethering the molecule to the surface while enabling free diffusion of the molecule. We ensure that each vesicle contains only a single RNA molecule, enabling single-molecule imaging. Upon dual-end labeling and encapsulation of the RNA of interest, smFRET measurements offer a dynamic and detailed view of RNA behavior.
Corrin Ring Modifications Reveal the Chemical and Spatial Requirements for the B12-btuB Riboswitch Interaction
Musiari, Anastasia; Reichenbach, María; Gallo, Sofia; Sigel, Roland K. O., Chem. Eur. J. 2024, 30 , e202401800.
The btuB riboswitch is a regulatory RNA sequence controlling gene expression of the outer membrane B12 transport protein BtuB by specifically binding coenzyme B12 (AdoCbl) as its natural ligand. The B12 sensing riboswitch class is known to accept various B12 derivatives, leading to a division into two riboswitch subclasses, dependent on the size of the apical ligand. Here we focus on the role of side chains b and e on affinity and proper recognition, i. e. correct structural switch of the btuB RNA, which belongs to the AdoCbl-binding class I. Chemical modification of these side chains disturbs crucial hydrogen bonds and/or electrostatic interactions with the RNA, its effect on both affinity and switching being monitored by in-line probing. Chemical modifications at sidechain b of vitamin B12 show larger effects indicating crucial B12-RNA interactions. When introducing the same modification to AdoCbl the influence of any side-chain modification tested is reduced. This renders the impact of the adenosyl-ligand for B12-btuB riboswitch recognition clearly beyond the known role in affinity.
FRET-guided modeling of nucleic acids
Steffen, Fabio D.; Cunha, Richard A.; Sigel, Roland K.O.; Börner, Richard, Nucleic Acids Res. 2024, 13, e59.
The functional diversity of RNAs is encoded in their innate conformational heterogeneity. The combination of single-molecule spectroscopy and computational modeling offers new attractive opportunities to map structural transitions within nucleic acid ensembles. Here, we describe a framework to harmonize single-molecule Förster resonance energy transfer (FRET) measurements with molecular dynamics simulations and de novo structure prediction. Using either all-atom or implicit fluorophore modeling, we recreate FRET experiments in silico, visualize the underlying structural dynamics and quantify the reaction coordinates. Using multiple accessible-contact volumes as a post hoc scoring method for fragment assembly in Rosetta, we demonstrate that FRET can be used to filter a de novo RNA structure prediction ensemble by refuting models that are not compatible with in vitro FRET measurement. We benchmark our FRET-assisted modeling approach on double-labeled DNA strands and validate it against an intrinsically dynamic manganese(II)-binding riboswitch. We show that a FRET coordinate describing the assembly of a four-way junction allows our pipeline to recapitulate the global fold of the riboswitch displayed by the crystal structure. We conclude that computational fluorescence spectroscopy facilitates the interpretability of dynamic structural ensembles and improves the mechanistic understanding of nucleic acid interactions.
Reply to: On the statistical foundation of a recent single molecule FRET benchmark
Götz, Markus; Barth, Anders; Bohr, Soren S.-R. et al., Nature Commun. 2024, 15, 3626.
In their ‘Matters Arising’ manuscript, Saurabh et al. discuss two issues related to single-molecule Förster resonance energy transfer (smFRET) experiments: the use of the Gaussian noise approximation and spectral crosstalk. Their arguments are based on simulations obtained with parameters that differ significantly from the typical conditions measured experimentally, and, thus, from the regime included in the original study (Götz et al.1). In addition, they make claims about our multi-lab blind study that we would like to rectify. In Table 1, we provide a list of specific statements made by Saurabh et al. with our respective explanations.
From Enigma to Revelation: Unravelling Biological Functions of Ubiquitous Small Ribozymes
Kienbeck, Kasimir; Malfertheiner, Lukas; Zelger-Paulus, Susann; Johannsen, Silke; von Mering, Christian; Sigel, Roland K. O., CHIMIA 2024, 78 , 200.
RNA, widely recognized as an information-carrier molecule, is capable of catalyzing essential biological processes through ribozymes. Despite their ubiquity, specific functions in a biological context and phenotypes based on the ribozymes' activity are often unknown. Here, we present the discovery of a subgroup of minimal HDV-like ribozymes, which reside 3' to viral tRNAs and appear to cleave the 3'-trailers of viral premature tRNA transcripts. This proposed tRNA-processing function is unprecedented for any ribozymes, thus, we designate this subgroup as theta ribozymes. Most theta ribozymes were identified in Caudoviricetes bacteriophages, the main constituent (>90%) of the mammalian gut virome. Intriguingly, our findings further suggest the involvement of theta ribozymes in the transition of certain bacteriophages between distinct genetic codes, thus possibly contributing to the phage lysis trigger. Our discovery expands the limited repertoire of biological functions attributed to HDV-like ribozymes and provides insights into the fascinating world of RNA catalysis.
Identification of HDV-like theta ribozymes involved in tRNA-based recoding of gut bacteriophages
Kienbeck, Kasimir; Malfertheiner, Lukas; Zelger-Paulus, Susann; Johannsen, Silke; von Mering, Christian; Sigel, Roland K O., Nature Commun. 2024, 15 , 1559.
Trillions of microorganisms, collectively known as the microbiome, inhabit our bodies with the gut microbiome being of particular interest in biomedical research. Bacteriophages, the dominant virome constituents, can utilize suppressor tRNAs to switch to alternative genetic codes (e.g., the UAG stop-codon is reassigned to glutamine) while infecting hosts with the standard bacterial code. However, what triggers this switch and how the bacteriophage manipulates its host is poorly understood. Here, we report the discovery of a subgroup of minimal hepatitis delta virus (HDV)-like ribozymes – theta ribozymes – potentially involved in the code switch leading to the expression of recoded lysis and structural phage genes. We demonstrate their HDV-like self-scission behavior in vitro and find them in an unreported context often located with their cleavage site adjacent to tRNAs, indicating a role in viral tRNA maturation and/or regulation. Every fifth associated tRNA is a suppressor tRNA, further strengthening our hypothesis. The vast abundance of tRNA-associated theta ribozymes – we provide 1753 unique examples – highlights the importance of small ribozymes as an alternative to large enzymes that usually process tRNA 3’-ends. Our discovery expands the short list of biological functions of small HDV-like ribozymes and introduces a previously unknown player likely involved in the code switch of certain recoded gut bacteriophages.
2023
Group II Introns: Highly Structured yet Dynamic
Ahunbay, Esra; Zelger-Paulus, Susann; Sigel, Roland K.O., CHIMIA 2023, 77, 235.
RNA splicing, the removal of introns and ligation of exons, is a crucial process during mRNA maturation. Group II introns are large ribozymes that self-catalyze their splicing, as well as their transposition. They are living fossils of spliceosomal introns and eukaryotic retroelements. The yeast mitochondrial Sc.ai5γ is the first identified and best-studied self-splicing group II intron. A combination of biochemical, biophysical, and computational tools enables studying its catalytic properties, structure, and dynamics, while also serving to develop new therapeutic and biotechnological tools. We survey the history of group II intron studies paralleling the trends in RNA methodology with Sc.ai5γ in the spotlight.
Organometallic Pillarplexes That Bind DNA 4-Way Holliday Junctions and Forks
Craig, James S.; Melidis, Larry; Williams, Hugo D.; Dettmer, Samuel J.; Heidecker, Alexandra A.; Altmann, Philipp J.; Guan, Shengyang, Campbell, Callum; Browning, Douglas F.; Sigel, Roland K. O.; Johannsen, Silke; Egan, Ross T.; Alikman, Brech; Casini, Angela; Pöthig, Alexander; Hannon, Michael J., J. Am. Chem. Soc. 2023, 145, 13570.
Holliday 4-way junctions are key to important biological DNA processes (insertion, recombination, and repair) and are dynamic structures that adopt either open or closed conformations, the open conformation being the biologically active form. Tetracationic metallo-supramolecular pillarplexes display aryl faces about a cylindrical core, an ideal structure to interact with open DNA junction cavities. Combining experimental studies and MD simulations, we show that an Au pillarplex can bind DNA 4-way (Holliday) junctions in their open form, a binding mode not accessed by synthetic agents before. Pillarplexes can bind 3-way junctions too, but their large size leads them to open up and expand that junction, disrupting the base pairing, which manifests in an increased hydrodynamic size and lower junction thermal stability. At high loading, they rearrange both 4-way and 3-way junctions into Y-shaped forks to increase the available junction-like binding sites. Isostructural Ag pillarplexes show similar DNA junction binding behavior but lower solution stability. This pillarplex binding contrasts with (but complements) that of metallo-supramolecular cylinders, which prefer 3-way junctions and can rearrange 4-way junctions into 3-way junction structures. The pillarplexes’ ability to bind open 4-way junctions creates exciting possibilities to modulate and switch such structures in biology, as well as in synthetic nucleic acid nanostructures. In human cells, the pillarplexes do reach the nucleus, with antiproliferative activity at levels similar to those of cisplatin. The findings provide a new roadmap for targeting higher-order junction structures using a metallo-supramolecular approach, as well as expanding the toolbox available to design bioactive junction binders into organometallic chemistry.
BOOK CHAPTER: Metabolite Regulation by Riboswitches - The Role of Metal Ions in Folding, Ligand Binding and Functionality
Reichenbach, Maria; Gallo, Sofia; Sigel, Roland K. O., In: Müller, J., & Lippert, B. (Eds), Modern Avenues in Metal-Nucleic Acid Chemistry (1st ed.) , 2023, CRC Press
Metal ions in association with RNA fulfill numerous functions and are vital for structure formation and functionality. K+ and Mg2+ are the most crucial, being the most abundant freely available metal ions in the cell. These ions not only shield the negative charges derived from the phosphate sugar backbone, but also allow the formation of highly complex, functional structures by mediating specific tertiary interactions. In ribozymes, specifically bound Mg2+ additionally promotes catalytic activity by metal ion-assisted RNA self-cleavage. In the context of riboswitches, metal ions take up further roles. Riboswitches are natural RNA aptamers involved in gene regulation by directly binding cellular metabolites or ions. In addition to their role in RNA folding, metal ions are often the key element for ligand binding allowing negatively charged moieties like phosphates, carboxylates, or even the single atomic fluoride to be recognized and bound by the RNA. Additionally, some riboswitch classes respond to metal ion-containing cofactors like molybdenum cofactor or cobalamins, while others are sensitive to single metal ions. For the latter, the RNA needs to discriminate between the correct metal ion and other, more abundant metal ions, a very difficult task to achieve in a cellular environment. For all these purposes riboswitches evolved elaborate binding pockets exploiting the different properties and binding strategies metal ions (and their ligands) can offer: size, softness/hardness, inner- and outer-sphere binding through an intricate H-bond pattern of coordinated water molecules as well as the inclusion of all functional moieties of the binding ligand. In this chapter, we discuss the different roles executed by metal ions for the proper functioning of riboswitches. Besides the general role during structure formation, we present different metal ion-dependent recognition modes, as well as metallo cofactor- and metal ion-binding riboswitches in more detail.
The structural features of the ligand-free moaA riboswitch and its ion-dependent folding
Amadei, Fabio; Reichenbach, María; Gallo, Sofia; Sigel, Roland K. O., J. Inorg. Biochem. 2023, 242, 112153.
Riboswitches are structural elements of mRNA involved in the regulation of gene expression by responding to specific cellular metabolites. To fulfil their regulatory function, riboswitches prefold into an active state, the so-called binding competent form, that guarantees metabolite binding and allows a consecutive refolding of the RNA. Here, we describe the folding pathway to the binding competent form as well as the ligand free structure of the moaA riboswitch of E. coli. This RNA proposedly responds to the molybdenum cofactor (Moco), a highly oxygen-sensitive metabolite, essential in the carbon and sulfur cycles of eukaryotes. K+- and Mg2+-dependent footprinting assays and spectroscopic investigations show a high degree of structure formation of this RNA already at very low ion-concentrations. Mg2+ facilitates additionally a general compaction of the riboswitch towards its proposed active structure. We show that this fold agrees with the earlier suggested secondary structure which included also a long-range tetraloop/tetraloop-receptor like interaction. Metal ion cleavage assays revealed specific Mg2+-binding pockets within the moaA riboswitch. These Mg2+ binding pockets are good indicators for the potential Moco binding site, since in riboswitches, Mg2+ was shown to be necessary to bind phosphate-carrying metabolites. The importance of the phosphate and of other functional groups of Moco is highlighted by binding assays with tetrahydrobiopterin, the reduced and oxygen-sensitive core moiety of Moco. We demonstrate that the general molecular shape of pterin by its own is insufficient for the recognition by the riboswitch.
2022
BOOK CHAPTER: Metal ion interactions with nucleic acids
Fazliji, Besim; Ferreira Rodrigues, Carla; Wang, Haibo; Sigel, Roland K. O., In: Reference Module in Chemistry, Molecular Sciences and Chemical Engineering , 2022,
This chapter focuses on the interaction of metal ions mainly with RNA. It is an update of the previous Chapter 3.21 in the 2nd Edition of Comprehensive Inorganic Chemistry II (2013) but focusing solely on RNA. Metal ions are key to folding, structure, and function of any nucleic acid. These interactions are generally of a weak and highly dynamic nature as they concern mostly K+ and Mg2+ in living organisms. Aside from the large excess of loosely bound ions for charge compensation, a network of inner-sphere and outer-sphere interactions holds more specifically bound ions in place. Hence, metal ion binding to larger RNAs is rather complicated and has many facets. After a few general considerations on the basic properties of metal ions and the potential coordination sites on the RNA, the thermodynamics of metal ion binding to RNA and known metal ion binding motifs in RNA are described. This is followed by today's knowledge on the role of metal ions in folding, dynamics, sensing, and/or catalysis of riboswitches and ribozymes, respectively, is summarized.
A blind benchmark of analysis tools to infer kinetic rate constants from single-molecule FRET trajectories
Götz, Markus; Barth, Anders; Bohr, Soren S.-R. et al., Nature Commun. 2022, 13, 5402.
Single-molecule FRET (smFRET) is a versatile technique to study the dynamics and function of biomolecules since it makes nanoscale movements detectable as fluorescence signals. The powerful ability to infer quantitative kinetic information from smFRET data is, however, complicated by experimental limitations. Diverse analysis tools have been developed to overcome these hurdles but a systematic comparison is lacking. Here, we report the results of a blind benchmark study assessing eleven analysis tools used to infer kinetic rate constants from smFRET trajectories. We test them against simulated and experimental data containing the most prominent difficulties encountered in analyzing smFRET experiments: different noise levels, varied model complexity, non-equilibrium dynamics, and kinetic heterogeneity. Our results highlight the current strengths and limitations in inferring kinetic information from smFRET trajectories. In addition, we formulate concrete recommendations and identify key targets for future developments, aimed to advance our understanding of biomolecular dynamics through quantitative experiment-derived models.
BOOK CHAPTER: "Single-Molecule Kinetic Studies of Nucleic Acids by Förster Resonance Energy Transfer”
Hadzic, Mélodie C. A. S.; Sigel, Roland K. O.; Börner, Richard, In: Steger G., Rosenbach H., Span I. (eds), DNAzymes. Methods in Molecular Biology , 2022, 2439 , Humana, New York, NY.
Single-molecule microscopy is often used to observe and characterize the conformational dynamics of nucleic acids (NA). Due to the large variety of NA structures and the challenges specific to single-molecule observation techniques, the data recorded in such experiments must be processed via multiple statistical treatments to finally yield a reliable mechanistic view of the NA dynamics. In this chapter, we propose a comprehensive protocol to analyze single-molecule trajectories in the scope of single-molecule Förster resonance energy transfer (FRET) microscopy. The suggested protocol yields the conformational states common to all molecules in the investigated sample, together with the associated conformational transition kinetics. The given model resolves states that are indistinguishable by their observed FRET signals and is estimated with 95% confidence using error calculations on FRET states and transition rate constants. In the end, a step-by-step user guide is given to reproduce the protocol with the Multifunctional Analysis Software to Handle single-molecule FRET data (MASH-FRET).
Magnesium(II)-ATP Complexes in 1-Ethyl-3-Methylimidazolium Acetate Solutions Characterized by 31Mg β-Radiation-Detected NMR Spectroscopy
McFadden, Ryan M. L.; Szunyogh, Dániel; Bravo-Frank, Nicholas; Chatzichristos, Aris; Dehn, Martin H.; Fujimoto, Derek; Jancsó, Attila; Johannsen, Silke; Kálomista, Ildikó; Karner, Victoria L.; Kiefl, Robert F.; Larsen, Flemming H.; Lassen, Jens; Levy, C. D. Philip; Li, Ruohong; McKenzie, Iain; McPhee, Hannah; Morris, Gerald D.; Pearson, Matthew R.; Sauer, Stephan P. A.; Sigel, Roland K. O.; Thulstrup, Peter W.; MacFarlane, W. Andrew; Hemmingsen, Lars; Stachura, Monika, Angew. Chem. Int. Ed. 2022, 61, e202207137
The complexation of MgII with adenosine 5′-triphosphate (ATP) is omnipresent in biochemical energy conversion, but is difficult to interrogate directly. Here we use the spin-urn:x-wiley:14337851:media:anie202207137:anie202207137-math-0001 β-emitter 31Mg to study MgII-ATP complexation in 1-ethyl-3-methylimidazolium acetate (EMIM-Ac) solutions using β-radiation-detected nuclear magnetic resonance (β-NMR). We demonstrate that (nuclear) spin-polarized 31Mg, following ion-implantation from an accelerator beamline into EMIM-Ac, binds to ATP within its radioactive lifetime before depolarizing. The evolution of the spectra with solute concentration indicates that the implanted 31Mg initially bind to the solvent acetate anions, whereafter they undergo dynamic exchange and form either a mono- (31Mg-ATP) or di-nuclear (31MgMg-ATP) complex. The chemical shift of 31Mg-ATP is observed up-field of 31MgMg-ATP, in accord with quantum chemical calculations. These observations constitute a crucial advance towards using β-NMR to probe chemistry and biochemistry in solution.
Coordination Chemistry of Nucleotides and Antivirally Active Acyclic Nucleoside Phosphonates, including Mechanistic Considerations
Sigel, Astrid; Sigel, Helmut; Sigel, Roland K. O., Molecules 2022, 27, 2625
Considering that practically all reactions that involve nucleotides also involve metal ions, it is evident that the coordination chemistry of nucleotides and their derivatives is an essential corner stone of biological inorganic chemistry. Nucleotides are either directly or indirectly involved in all processes occurring in Nature. It is therefore no surprise that the constituents of nucleotides have been chemically altered—that is, at the nucleobase residue, the sugar moiety, and also at the phosphate group, often with the aim of discovering medically useful compounds. Among such derivatives are acyclic nucleoside phosphonates (ANPs), where the sugar moiety has been replaced by an aliphatic chain (often also containing an ether oxygen atom) and the phosphate group has been replaced by a phosphonate carrying a carbon–phosphorus bond to make the compounds less hydrolysis-sensitive. Several of these ANPs show antiviral activity, and some of them are nowadays used as drugs. The antiviral activity results from the incorporation of the ANPs into the growing nucleic acid chain—i.e., polymerases accept the ANPs as substrates, leading to chain termination because of the missing 3′-hydroxyl group. We have tried in this review to describe the coordination chemistry (mainly) of the adenine nucleotides AMP and ATP and whenever possible to compare it with that of the dianion of 9-[2-(phosphonomethoxy)ethyl]adenine (PMEA2− = adenine(N9)-CH2-CH2-O-CH2-PO23) [or its diphosphate (PMEApp4−)] as a representative of the ANPs. Why is PMEApp4− a better substrate for polymerases than ATP4−? There are three reasons: (i) PMEA2− with its anti-like conformation (like AMP2−) fits well into the active site of the enzyme. (ii) The phosphonate group has an enhanced metal ion affinity because of its increased basicity. (iii) The ether oxygen forms a 5-membered chelate with the neighboring phosphonate and favors thus coordination at the Pα group. Research on ANPs containing a purine residue revealed that the kind and position of the substituent at C2 or C6 has a significant influence on the biological activity. For example, the shift of the (C6)NH2 group in PMEA to the C2 position leads to 9-[2-(phosphonomethoxy)ethyl]-2-aminopurine (PME2AP), an isomer with only a moderate antiviral activity. Removal of (C6)NH2 favors N7 coordination, e.g., of Cu2+, whereas the ether O atom binding of Cu2+ in PMEA facilitates N3 coordination via adjacent 5- and 7-membered chelates, giving rise to a Cu(PMEA)cl/O/N3 isomer. If the metal ions (M2+) are M(α,β)-M(γ)-coordinated at a triphosphate chain, transphosphorylation occurs (kinases, etc.), whereas metal ion binding in a M(α)-M(β,γ)-type fashion is relevant for polymerases. It may be noted that with diphosphorylated PMEA, (PMEApp4−), the M(α)-M(β,γ) binding is favored because of the formation of the 5-membered chelate involving the ether O atom (see above). The self-association tendency of purines leads to the formation of dimeric [M2(ATP)]2(OH)− stacks, which occur in low concentration and where one half of the molecule undergoes the dephosphorylation reaction and the other half stabilizes the structure—i.e., acts as the “enzyme” by bridging the two ATPs. In accord herewith, one may enhance the reaction rate by adding AMP2− to the [Cu2(ATP)]2(OH)− solution, as this leads to the formation of mixed stacked Cu3(ATP)(AMP)(OH)− species, in which AMP2− takes over the structuring role, while the other “half” of the molecule undergoes dephosphorylation. It may be added that Cu3(ATP)(PMEA) or better Cu3(ATP)(PMEA)(OH)− is even a more reactive species than Cu3(ATP)(AMP)(OH)−. – The matrix-assisted self-association and its significance for cell organelles with high ATP concentrations is summarized and discussed, as is, e.g., the effect of tryptophanate (Trp−), which leads to the formation of intramolecular stacks in M(ATP)(Trp)3− complexes (formation degree about 75%). Furthermore, it is well-known that in the active-site cavities of enzymes the dielectric constant, compared with bulk water, is reduced; therefore, we have summarized and discussed the effect of a change in solvent polarity on the stability and structure of binary and ternary complexes: Opposite effects on charged O sites and neutral N sites are observed, and this leads to interesting insights.
BOOK CHAPTER: "Chemical Dual End-Labeling of Large Ribozymes”
Ahunbay, Esra; Steffen Fabio D.; Zelger-Paulus, Susann; Sigel, Roland K. O., In: Steger G., Rosenbach H., Span I. (eds), DNAzymes. Methods in Molecular Biology , 2022, 2439 , Humana, New York, NY.
Fast and efficient site-specific labeling of long RNAs is one of the main bottlenecks limiting distance measurements by means of Förster resonance energy transfer (FRET) or electron paramagnetic resonance (EPR) spectroscopy. Here, we present an optimized protocol for dual end-labeling with different fluorophores at the same time meeting the restrictions of highly labile and degradation-sensitive RNAs. We describe in detail the dual-labeling of a catalytically active wild-type group II intron as a typical representative of long functional RNAs. The modular procedure chemically activates the 5′-phosphate and the 3′-ribose for bioconjugation with a pair of fluorophores, as shown herein, or with spin labels. The mild reaction conditions preserve the structural and functional integrity of the biomacromolecule and results in covalent, dual-labeled RNA in its pre-catalytic state in yields suitable for both ensemble and single-molecule FRET experiments.
2021
FRETraj: Integrating Single-Molecule Spectroscopy with Molecular Dynamics
Steffen, Fabio D.; Sigel, Roland K. O.; Börner, Richard, Bioinformatics 2021,
Puf6 Primes 60S Pre-Ribosome Nuclear Export at Low Temperature
Gerhardy, Stefan; Oborská-Oplová, Michaela; Gillet, Ludovic; Börner, Richard; van Nues, Rob; Leitner, Alexander; Michel, Erich; Petkowski, Janusz J.; Granneman, Sander; Sigel, Roland K. O.; Aebersold, Ruedi; Panse, Vikram G., Nature Commun. 2021, 12 , 4696.
Productive ribosomal RNA (rRNA) compaction during ribosome assembly necessitates establishing correct tertiary contacts between distant secondary structure elements. Here, we quantify the response of the yeast proteome to low temperature (LT), a condition where aberrant mis-paired RNA folding intermediates accumulate. We show that, at LT, yeast cells globally boost production of their ribosome assembly machinery. We find that the LT-induced assembly factor, Puf6, binds to the nascent catalytic RNA-rich subunit interface within the 60S pre-ribosome, at a site that eventually loads the nuclear export apparatus. Ensemble Förster resonance energy transfer studies show that Puf6 mimics the role of Mg2+ to usher a unique long-range tertiary contact to compact rRNA. At LT, puf6 mutants accumulate 60S pre-ribosomes in the nucleus, thus unveiling Puf6-mediated rRNA compaction as a critical temperature-regulated rescue mechanism that counters rRNA misfolding to prime export competence.
Screening for potential interaction partners with surface plasmon resonance imaging coupled to MALDI mass spectrometry
Anders, Ulrike; Gulotti-Georgieva, Maya; Zelger-Paulus, Susann; Hibti, Fatima-Ezzahra; Frydman, Chiraz; Suckau, Detlev; Sigel, Roland K. O.; Zenobi, Renato, Anal. Biochem. 2021, 624 , 114195.
We coupled SPR imaging (SPRi) with matrix-assisted laser desorption/ionization mass spectrometry (MALDI MS) to identify new potential RNA binders. Here, we improve this powerful method, especially by optimizing the proteolytic digestion (type of reducing agent, its concentration, and incubation time), to work with complex mixtures, specifically a lysate of the rough mitochondrial fraction from yeast. The advantages of this hyphenated method compared to column-based or separate analyses are (i) rapid and direct visual readout from the SPRi array, (ii) possibility of high-throughput analysis of different interactions in parallel, (iii) high sensitivity, and (iv) no sample loss or contamination due to elution or micro-recovery procedures. The model system used is a catalytically active RNA (group IIB intron from Saccharomyces cerevisiae, Sc.ai5γ) and its cofactor Mss116. The protein supports the RNA folding process and thereby the subsequent excision of the intronic RNA from the coding part. Using the novel approach of coupling SPR with MALDI MS, we report the identification of potential RNA-binding proteins from a crude yeast mitochondrial lysate in a non-targeted approach. Our results show that proteins other than the well-known cofactor Mss116 interact with Sc.ai5γ (Dbp8, Prp8, Mrp13, and Cullin-3), suggesting that the intron folding and splicing are regulated by more than one cofactor in vivo.
2020
Metal ions and sugar puckering balance single-molecule kinetic heterogeneity in RNA and DNA tertiary contacts
Steffen, Fabio D.; Khier, Mokrane; Kowerko, Danny; Cunha, Richard A.; Börner, Richard; Sigel, Roland K. O., Nat. Commun. 2020, 11 , 104.
The fidelity of group II intron self-splicing and retrohoming relies on long-range tertiary interactions between the intron and its flanking exons. By single-molecule FRET, we explore the binding kinetics of the most important, structurally conserved contact, the exon and intron binding site 1 (EBS1/IBS1). A comparison of RNA-RNA and RNA-DNA hybrid contacts identifies transient metal ion binding as a major source of kinetic heterogeneity which typically appears in the form of degenerate FRET states. Molecular dynamics simulations suggest a structural link between heterogeneity and the sugar conformation at the exon-intron binding interface. While Mg2+ ions lock the exon in place and give rise to long dwell times in the exon bound FRET state, sugar puckering alleviates this structural rigidity and likely promotes exon release. The interplay of sugar puckering and metal ion coordination may be an important mechanism to balance binding affinities of RNA and DNA interactions in general.
2019
The Bioinorganic Periodic Table
Freisinger, Eva; Sigel, Roland K. O., CHIMIA 2019, 73 , 185-193.
Life depends on metals. While carbon, in terms of abundance and versatility, is considered THE element of life, the vast variety and diversity of the chemistry taking place in living organisms could not be achieved without metal ions. More than twenty metals are found in the human body, most of them being essential, some beneficial, and for others it is still unknown what role they might fulfil in a living cell. Here we give a short introduction into the bioinorganic world of the periodic table, providing just a few examples of key metals for life and aiming to give a flavour to gain further insights into this exciting field of inorganic chemistry at the intersection to the life sciences.
NMR solution structure of tricyclo-DNA containing duplexes: insight into enhanced thermal stability and nuclease resistance
Istrate, Andrei; Johannsen, Silke; Istrate, Alena; Sigel, Roland K. O.; Leumann, Christian J., Nucleic Acids Res. 2019, 47 , 4872-4882.
Tc-DNA is a conformationally constrained oligonucleotide analogue which shows significant increase in thermal stability when hybridized with RNA, DNA or tc-DNA. Remarkably, recent studies revealed that tc-DNA antisense oligonucleotides (AO) hold great promise for the treatment of Duchenne muscular dystrophy and spinal muscular atrophy. To date, no high-resolution structural data is available for fully modified tc-DNA duplexes and little is known about the origins of their enhanced thermal stability. Here, we report the structures of a fully modified tc-DNA oligonucleotide paired with either complementary RNA, DNA or tc-DNA. All three investigated duplexes maintain a right-handed helical structure with Watson-Crick base pairing and overall geometry intermediate between A- and B-type, but closer to A-type structures. All sugars of the tc-DNA and RNA residues adopt a North conformation whereas the DNA deoxyribose are found in a South-East-North conformation equilibrium. The conformation of the tc-DNA strand in the three determined structures is nearly identical and despite the different nature and local geometry of the complementary strand, the overall structures of the examined duplexes are very similar suggesting that the tc-DNA strand dominates the duplex structure.
Concerted dynamics of metallo-base pairs in an A/B-form helical transition
Schmidt, Olivia P.; Jurt, Simon; Johannsen, Silke; Karimi, Ashkan; Sigel, Roland K.O.; Luedtke, Nathan W., Nat. Commun. 2019, 10 , 4818.
Metal-mediated base pairs expand the repertoire of nucleic acid structures and dynamics. Here we report solution structures and dynamics of duplex DNA containing two all-natural C-HgII-T metallo base pairs separated by six canonical base pairs. NMR experiments reveal a 3:1 ratio of well-resolved structures in dynamic equilibrium. The major species contains two (N3)T-HgII-(N3)C base pairs in a predominantly B-form helix. The minor species contains (N3)T-HgII-(N4)C base pairs and greater A-form characteristics. Ten-fold different 1J coupling constants (15N,199Hg) are observed for (N3)C-HgII (114 Hz) versus (N4)C-HgII (1052 Hz)~connectivities, reflecting differences in cytosine ionization and metal-bonding strengths. Dynamic interconversion between the two types of C-HgII-T base pairs are coupled to a global conformational exchange between the helices. These observations inspired the design of a repetitive DNA sequence capable of undergoing a global B-to-A-form helical transition upon adding HgII, demonstrating that C-HgII-T has unique switching potential in DNA-based materials and devices.
Stick, Flick, Click: DNA-guided Fluorescent Labeling of Long RNA for Single-molecule FRET
Steffen, Fabio D.; Börner, Richard; Freisinger, Eva; Sigel, Roland K. O., CHIMIA 2019, 73 , 257-261.
Exploring the spatiotemporal dynamics of biomolecules on a single-molecule level requires innovative ways to make them spectroscopically visible. Fluorescence resonance energy transfer (FRET) uses a pair of organic dyes as reporters to measure distances along a predefined biomolecular reaction coordinate. For this nanoscopic ruler to work, the fluorescent labels need to be coupled onto the molecule of interest in a bioorthogonal and site-selective manner. Tagging large non-coding RNAs with single-nucleotide precision is an open challenge. Here we summarize current strategies in labeling riboswitches and ribozymes for fluorescence spectroscopy and FRET in particular. A special focus lies on our recently developed, DNA-guided approach that inserts two fluorophores through a stepwise process of templated functionality transfer and click chemistry.
2018
Simulations of camera-based single-molecule fluorescence experiments
Börner, Richard; Kowerko, Danny; Hadzic, Mélodie C. A. S.; König, Sebastian L. B.; Ritter, Marc; Sigel, Roland K. O., PLoS One 2018, 13 , e0195277.
Single-molecule microscopy has become a widely used technique in (bio)physics and (bio)chemistry. A popular implementation is single-molecule Förster Resonance Energy Transfer (smFRET), for which total internal reflection fluorescence microscopy is frequently combined with camera-based detection of surface-immobilized molecules. Camera-based smFRET experiments generate large and complex datasets and several methods for video processing and analysis have been reported. As these algorithms often address similar aspects in video analysis, there is a growing need for standardized comparison. Here, we present a Matlab-based software (MASH-FRET) that allows for the simulation of camera-based smFRET videos, yielding standardized data sets suitable for benchmarking video processing algorithms. The software permits to vary parameters that are relevant in cameras-based smFRET, such as video quality, and the properties of the system under study. Experimental noise is modeled taking into account photon statistics and camera noise. Finally, we survey how video test sets should be designed to evaluate currently available data analysis strategies in camera-based sm fluorescence experiments. We complement our study by pre-optimizing and evaluating spot detection algorithms using our simulated video test sets.
Specific phosphorothioate substitution within domain 6 of a group II intron ribozyme leads to changes in local structure and metal ion binding
Erat, Michèle C.; Besic-Gyenge, Emina; Oberhuber, Michael; Johannsen, Silke; Sigel, Roland K. O., J. Biol. Inorg. Chem. 2018, 23 , 167-177.
Group II introns are large self-splicing ribozymes that require high amounts of monovalent and divalent metal ions for folding and catalysis under in vitro conditions. Domain 6 of these ribozymes contains a highly conserved adenosine whose 2′-OH acts as a nucleophile during self-cleavage via the branching pathway. We have previously suggested a divalent metal ion that binds to the major groove at the GU wobble pair above the branch-A in a minimal, but active branch domain construct (D6–27) from the yeast mitochondrial intron Sc.ai5γ. Here we characterize metal ion binding to the phosphate oxygens at the branch site. In vitro transcription yielded a D6–27 construct where all RP oxygens of the uridine phosphate groups are replaced by sulfur (α-thio-D6–27). We determined its NMR structure, the second RNA-only structure containing thiophosphate groups. [31P] resonances were assigned and chemical shift changes monitored upon titration with Cd2+. In addition, the two uridines flanking the branch-point, U19 and U21 were specifically thioated by chemical synthesis (thio-U19-D6–27 and thio-U19/U21-D6–27), enabling us to study Cd2+ binding at the RP-, as well as the SP- position of the corresponding phosphate oxygens. Our studies reveal that both non-bridging phosphate oxygens of U19 are involved in metal ion coordination, whereas only the major groove phosphate oxygen of U21 is influenced. Together with NOE data of a hexaamminecobalt(III) titration, this suggests a single metal ion binding site at the GU wobble pair above the branch point in the major groove of D6 of this group II intron ribozyme.
Covalent and non-covalent binding of platinated vitamin B 12 -derivatives to a B 12 responsive riboswitch
Gallo, Sofia; Sigel, Roland K. O., Inorg. Chim. Acta 2018, 472 , 214-220.
The B12 responding btuB riboswitch is a short, non-coding RNA sequence involved in the gene-regulation of an outer membrane B12-transport protein in E. coli. This RNA is characterized by its selective high-affinity binding to coenzyme B12 and by the structural rearrangement it undergoes upon this interaction. Due to their involvement in (mostly) bacterial gene regulation, the btuB riboswitch as well as the further about twenty known classes of riboswitches received much attention in recent years. In case of the btuB riboswitch, the light sensitivity of its ligand, coenzyme B12, poses one of the greater challenges for its investigation. Vitamin B12 derivatives carrying a cyanide-bridged platinum(II) moiety offer an ideal strategy for the design of light stable coenzyme B12 analogs. Developed by Alberto and coworkers in 2005 these conjugates provide the possibility to coordinate an additional nucleobase making them potential coenzyme B12 mimics. However, although these derivatives show a high structural similarity to coenzyme B12, their functionality might differ and offers the opportunity to develop new classes of antibiotic agents. Especially the platinum(II)-complex could interact with RNA in an unexpected way, which is the main question addressed in the work presented here. We characterized the binding of three vitamin B12-platinum(II) complexes to two different RNAs: the B12-specific btuB riboswitch and the short RNA D1-45, which is a non-B12-binder. cisPt(II)VitB12+ (1) and enPt(II)VitB12+ (2) carry both a labile chloride ligand at the platinum(II) moiety whereas dienPt(II)VitB122+ (3), that carries no labile chloride ligand anymore, can be considered chemically inert under the applied conditions. Complexes 1 and 2 covalently bind to both RNAs as monitored by band-shift assays. In the case of the short D1-45 the shifted bands are well separated and were further analyzed by MALDI-MS proving the covalent interaction. Complex 3 is unable to covalently bind any of the two RNAs, which proves the general stability of the Pt(II) complex. However, the presence of this moiety has a dramatic influence on the binding property towards the B12-sensing riboswitch, as was observed in in-line probing assays by comparison to the natural ligand coenzyme B12.
Reliable State Identification and State Transition Detection in Fluorescence Intensity-Based Single-Molecule Förster Resonance Energy-Transfer Data
Hadzic, Mélodie C. A. S.; Börner, Richard; König, Sebastian L. B.; Kowerko, Danny; Sigel, Roland K. O., J. Phys. Chem. B 2018, epub , online.
Single-molecule Förster resonance energy transfer (smFRET) is a powerful technique to probe biomolecular structure and dynamics. A popular implementation of smFRET consists of recording fluorescence intensity time traces of surface-immobilized, chromophore-tagged molecules. This approach generates large and complex data sets, the analysis of which is to date not standardized. Here, we address a key challenge in smFRET data analysis: the generation of thermodynamic and kinetic models that describe with statistical rigor the behavior of FRET trajectories recorded from surface-tethered biomolecules in terms of the number of FRET states, the corresponding mean FRET values, and the kinetic rates at which they interconvert. For this purpose, we first perform Monte Carlo simulations to generate smFRET trajectories, in which a relevant space of experimental parameters is explored. Then, we provide an account on current strategies to achieve such model selection, as well as a quantitative assessment of their performances. Specifically, we evaluate the performance of each algorithm (change-point analysis, STaSI, HaMMy, vbFRET, and ebFRET) with respect to accuracy, reproducibility, and computing time, which yields a range of algorithm-specific referential benchmarks for various data qualities. Data simulation and analysis were performed with our MATLAB-based multifunctional analysis software for handling smFRET data (MASH-FRET).
Optimal molecular crowding accelerates group II intron folding and maximizes catalysis
Paudel, Bishnu P.; Fiorini, Erica; Börner, Richard; Sigel, Roland K O; Rueda, David S., Proc. Natl. Acad. Sci. U. S. A. 2018, 115 , 11917-11922.
Unlike in vivo conditions, group II intron ribozymes are known to require high magnesium(II) concentrations ([Mg2+]) and high temperatures (42 °C) for folding and catalysis in vitro. A possible explanation for this difference is the highly crowded cellular environment, which can be mimicked in vitro by macromolecular crowding agents. Here, we combined bulk activity assays and single-molecule Förster Resonance Energy Transfer (smFRET) to study the influence of polyethylene glycol (PEG) on catalysis and folding of the ribozyme. Our activity studies reveal that PEG reduces the [Mg2+] required, and we found an {\textquotedbl}optimum{\textquotedbl} [PEG] that yields maximum activity. smFRET experiments show that the most compact state population, the putative active state, increases with increasing [PEG]. Dynamic transitions between folded states also increase. Therefore, this study shows that optimal molecular crowding concentrations help the ribozyme not only to reach the native fold but also to increase its in vitro activity to approach that in physiological conditions.
Metal ion complexes of nucleoside phosphorothioates reflecting the ambivalent properties of lead( ii )
Sigel, Astrid; Operschall, Bert P.; Sigel, Roland K. O.; Sigel, Helmut, New J. Chem. 2018, 42 , 7551-7559.
This Perspective outlines the coordinating properties of lead(II), to some extent in comparison with related metal ions like Ca2+, Zn2+ or Cd2+. It is worth noting that the affinity of Pb2+ towards phosphate residues corresponds to that of Cu2+. Furthermore, the binding tendency of Pb2+ towards thiophosphate groups as present in methyl thiophosphate (MeOPS2−) or uridine 5′-O-thiomonophosphate (UMPS2−) is compared with that of the parent ligands, that is, methyl phosphate (CH3OPO32−) and uridine 5′-monophosphate (UMP2−). The replacement of an O by a S atom makes the monoprotonated thiophosphate group considerably more acidic [compared to ROP(O)2−(OH)], but at the same time its affinity for Pb2+ increases tremendously: more than 99{\%} of Pb2+ is S-bound. This is very different if the coordinating properties of uridylyl-(5′→3′)-[5′]-uridylate (pUpU3−) and P-thiouridylyl-(5′→3′)-[5′]-uridylate (pUp(S)U3−) are compared. The phosphate-coordinated Pb2+ forms a 10-membered chelate with one of the two terminal O atoms of the phosphodiester linkage, which reaches a formation degree of about 90{\%} in Pb(pUpU)−. However, in Pb(pUp(S)U)− the formation degree of the chelate is reduced to about half in accordance with the fact that now only one terminal O atom is available in the thiophosphate diester bridge, that is, Pb2+ coordinates to this O showing no affinity for S in ROP(O)(S)−OR′. These observations are ascribed to the properties of the Pb2+ lone pair, which shapes the Pb2+ coordination sphere; its role is discussed further in this Perspective and a caveat is made regarding Pb2+ binding to a thiophosphate diester linkage.
Site-specific dual-color labeling of long RNAs for single-molecule spectroscopy
Zhao, Meng; Steffen, Fabio D.; Börner, Richard; Schaffer, Michelle F.; Sigel, Roland K. O.; Freisinger, Eva, Nucleic Acids Res. 2018, 46 , e13.
Labeling of long RNA molecules in a site-specific yet generally applicable manner is integral to many spectroscopic applications. Here we present a novel covalent labeling approach that is site-specific and scalable to long intricately folded RNAs. In this approach, a custom-designed DNA strand that hybridizes to the RNA guides a reactive group to target a preselected adenine residue. The functionalized nucleotide along with the concomitantly oxidized 3'-terminus can subsequently be conjugated to two different fluorophores via bio-orthogonal chemistry. We validate this modular labeling platform using a regulatory RNA of 275 nucleotides, the btuB riboswitch of Escherichia coli, demonstrate its general applicability by modifying a base within a duplex, and show its site-selectivity in targeting a pair of adjacent adenines. Native folding and function of the RNA is confirmed on the single-molecule level by using FRET as a sensor to visualize and characterize the conformational equilibrium of the riboswitch upon binding of its cofactor adenosylcobalamin. The presented labeling strategy overcomes size and site constraints that have hampered routine production of labeled RNA that are beyond 200 nt in length.
2017
G-quadruplex DNA targeted metal complexes acting as potential anticancer drugs
Cao, Qian; Li, Yi; Freisinger, Eva; Qin, Peter Z.; Sigel, Roland K. O.; Mao, Zong-Wan, Inorg. Chem. Front. 2017, 4 , 10-32.
Although cisplatin and its analogues have been widely utilized as anticancer metallodrugs in clinics, their serious side effects and damage to normal tissues cannot be avoided because cisplatin kills cancer cells by attacking genomic DNA. Thus the design of metallodrugs possessing different actions of anticancer mechanism is promising. G-quadruplex nucleic acid, which is formed by self-assembly of guanine-rich nucleic acid sequences, has recently been considered as an attractive target for anticancer drug design. The basic unit of a G-quadruplex is a G-quartet, a planar motif generated from four guanine residues pairing together through Hoogsteen like hydrogen bonds. DNA G-quadruplex (G4) structures exist in the chromosomal telomeric sequences and the promoter regions of numerous genes, including oncogenetic promoters. Formation of G4 structures within the 3’-overhang of telomeric DNA can inhibit the telomerase activity, which is silent in normal cells but up-regulated in most cancer cells, thus significantly shortening telomeres and preventing cancer cell proliferation and immortalization. Intramolecular G4 structures formed within the oncogene promoter regions can effectively inhibit oncogenen transcription and expression. Thus rational design of small molecular ligands to selectively interact, stabilize or cleave G4 structures is a promising strategy for developing potent anti-cancer drugs with selective toxicity towards cancer cells over normal ones. This review will highlight the recent development of G4-interacting metal complexes, termed G4-ligands, discussing their binding modes with G-quadruplex DNA and their potential to serve as anticancer drugs in the medical field.
Tb3+-Cleavage Assays Reveal Specific Mg2+ Binding Sites Necessary to Pre-fold the btuB Riboswitch for AdoCbl Binding
Choudhary, Pallavi K.; Gallo, Sofia; Sigel, Roland K. O., Front. Chem. 2017, 5 , 598.
Riboswitches are RNA elements that bind specific metabolites in order to regulate the gene expression involved in controlling the cellular concentration of the respective molecule or ion. Ligand recognition is mostly facilitated by Mg2+ mediated pre-organization of the riboswitch to an active tertiary fold. To predict these specific Mg2+ induced tertiary interactions of the btuB riboswitch from E. coli, we here report Mg2+ binding pockets in its aptameric part in both, the ligand-free and the ligand-bound form. An ensemble of weak and strong metal ion binding sites distributed over the entire aptamer was detected by terbium(III) cleavage assays, Tb3+ being an established Mg2+ mimic. Interestingly many of the Mn+ (n = 2 or 3) binding sites involve conserved bases within the class of coenzyme B12-binding riboswitches. Comparison with the published crystal structure of the coenzyme B12 riboswitch of S. thermophilum aided in identifying a common set of Mn+ binding sites that might be crucial for tertiary interactions involved in the organization of the aptamer. Our results suggest that Mn+ binding at strategic locations of the btuB riboswitch indeed facilitates the assembly of the binding pocket needed for ligand recognition. Binding of the specific ligand, coenzyme B12 (AdoCbl), to the btuB aptamer does however not lead to drastic alterations of these Mn+ binding cores, indicating the lack of a major rearrangement within the three-dimensional structure of the RNA. This finding is strengthened by Tb3+ mediated footprints of the riboswitch’s structure in its ligand-free and ligand-bound state indicating that AdoCbl indeed induces local changes rather than a global structural rearrangement.
Influence of pH and Mg(ii) on the catalytic core domain 5 of a bacterial group II intron
Pechlaner, Maria; Domínguez-Martín, Alicia; Sigel, Roland K. O., Dalton Trans. 2017, 46 , 3989-3995.
RNA molecules fold into complex structures that allow them to perform specific functions. To compensate the relative lack of diversity of functional groups within nucleotides, metal ions work as crucial co-factors. In addition, shifted pKas are observed in RNA, enabling acid-base reactions at ambient pH. The central catalytic domain 5 (D5) hairpin of the Azotobacter vinelandii group II intron undergoes both metal ion binding and pH dependence, presumably playing an important functional role in the ribozyme’s reaction. By NMR spectroscopy we have here characterized the metal ion binding sites and affinities for the hairpin’s internal G-A mismatch, bulge, and pentaloop. The influence of Mg(ii) and pH on the local conformation of the catalytically crucial region is also explored by fluorescence spectroscopy.
2016
Studying metal ion binding properties of a three-way junction RNA by heteronuclear NMR
Bartova, Simona; Pechlaner, Maria; Donghi, Daniela; Sigel, Roland K. O., J. Biol. Inorg. Chem. 2016, 21 , 319-328.
Self-splicing group II introns are highly structured RNA molecules, containing a characteristic secondary and catalytically active tertiary structure, which is formed only in the presence of Mg(II). Mg(II) initiates the first folding step governed by the κζ element within domain 1 (D1κζ). We recently solved the NMR structure of D1κζ derived from the mitochondrial group II intron ribozyme Sc.ai5γ and demonstrated that Mg(II) is essential for its stabilization. Here, we performed a detailed multinuclear NMR study of metal ion interactions with D1κζ, using Cd(II) and cobalt(III)hexammine to probe inner- and outer-sphere coordination of Mg(II) and thus to better characterize its binding sites. Accordingly, we mapped (1)H, (15)N, (13)C, and (31)P spectral changes upon addition of different amounts of the metal ions. Our NMR data reveal a Cd(II)-assisted macrochelate formation at the 5’-end triphosphate, a preferential Cd(II) binding to guanines in a helical context, an electrostatic interaction in the ζ tetraloop receptor and various metal ion interactions in the GAAA tetraloop and κ element. These results together with our recently published data on Mg(II) interaction provide a much better understanding of Mg(II) binding to D1κζ, and reveal how intricate and complex metal ion interactions can be.
Metal ion binding to an RNA internal loop
Bartova, Simona; Alberti, Elena; Sigel, Roland K. O.; Donghi, Daniela, Inorg. Chim. Acta 2016, 452 , 104-110.
Studying the interaction of metal ions with RNA is challenging because of the fast dynamics of the system and the intricate interplay between structural and functional roles of metal ions. NMR spectroscopy is an exceptional tool to investigate such interactions in solution and allows for a detailed description of both metal ion binding sites and binding modes in complex and dynamic RNA structures. We recently applied heteronuclear NMR to study the metal ion binding properties of a three-way junction RNA (D1jf) which plays an important role in group II intron splicing, and observed metal ion binding in both j and f regions of the construct. Here we concentrate in more detail on the f region (D1f) using NMR to investigate the interaction with Mg(II), Cd(II) and cobalt(III)hexammine. Our data confirm Cd(II) induced macrochelate formation at the 50-end triphosphate, suggest an overall similar behaviour for the two divalent metal ions, but with much clearer changes in chemical shifts upon Cd(II) addition, and reveal only little changes upon cobalt(III)hexammine addition, allowing to discriminate between inner- and outer-sphere binding. Moreover, we observed distinct differences when we titrated the sample with Cd(II) in the presence of either KCl or KClO4 as background monovalent salt.
Metal ion induced heterogeneity in RNA folding studied by smFRET - Review
Börner, Richard; Kowerko, Danny; Guiset Miserachs, Helena; Schaffer, Michelle F.; Sigel, Roland K. O., Coord. Chem. Rev. 2016, 327-328 , 123-142.
More than two decades of investigating nucleic acids and ribonucleic acids (RNA) using single molecule Förster resonance energy transfer (smFRET) have passed. It turned out that sample heterogeneity in structure and function of RNA molecules as well as folding intermediates, kinetic subpopulations, and interconversion rates of conformational states of RNA biomolecules, all of which are usually hidden in ensemble type experiments, are often observed characteristics. Besides proteins, metal ions play a crucial role in RNA folding and dynamics, as well as RNA/RNA or RNA/DNA interactions. RNA molecules form discrete conformational intermediates before reaching the native three-dimensional fold, whereby metal ions guide the folding pathway by changing the energetic barriers between local and global minima in the energy landscape. Here we review recent advances in the characterization of the role of metal ions in folding and function of nucleic acid structures by means of smFRET. Subsequently, the workflow of smFRET data analysis is described and exemplified by the metal ion-depending folding and dynamics of the group IIB intron from Saccharomyces cerevisiae and RNA–RNA binding kinetics of this ribozyme's 5'-splice site formation.
Sequence-Specific Post-Synthetic Oligonucleotide Labeling for Single-Molecule Fluorescence Applications
Egloff, David; Oleinich, Igor A.; Zhao, Meng; König, Sebastian L. B.; Sigel, Roland K. O.; Freisinger, Eva, ACS Chem. Biol. 2016, 11 , 2558-2567.
The sequence-specific fluorescence labeling of nucleic acids is a prerequisite for various methods including single-molecule Förster resonance energy transfer (smFRET) for the detailed study of nucleic acid folding and function. Such nucleic acid derivatives are commonly obtained by solid-phase methods; however, yields decrease rapidly with increasing length and restrict the practicability of this approach for long strands. Here, we report a new labeling strategy for the postsynthetic incorporation of a bioorthogonal group into single stranded regions of both DNA and RNA of unrestricted length. A 12-alkyne-etheno-adenine modification is sequence-selectively formed using DNA-templated synthesis, followed by conjugation of the fluorophore Cy3 via a copper-catalyzed azide-alkyne cycloaddition (CuAAC). Evaluation of the labeled strands in smFRET measurements shows that the strategy developed here has the potential to be used for the study of long functional nucleic acids by (single-molecule) fluorescence or other methods. To prove the universal use of the method, its application was successfully extended to the labeling of a short RNA single strand. As a proof-of-concept, also the labeling of a large RNA molecule in form of a 633 nucleotide long construct derived from the Saccharomyces cerevisiae group II intron Sc.ai5γ was performed, and covalent attachment of the Cy3 fluorophore was shown with gel electrophoresis.
Distinct differences in metal ion specificity of RNA and DNA G-quadruplexes
Guiset Miserachs, Helena; Donghi, Daniela; Börner, Richard; Johannsen, Silke; Sigel, Roland K. O., J. Biol. Inorg. Chem. 2016, 21 , 975-986.
RNA G-quadruplexes, as their well-studied DNA analogs, require the presence of cations to fold and remain stable. This is the first comprehensive study on the interaction of RNA quadruplexes with metal ions. We investigated the formation and stability of two highly conserved and biologically relevant RNA quadruplex-forming sequences (24nt-TERRA and 18nt-NRAS) in the presence of several monovalent and divalent metal ions, namely Li(+), Na(+), K(+), Rb(+), Cs(+), NH4(+), Mg(2+), Ca(2+), Sr(2+), and Ba(2+). Circular dichroism was used to probe the influence of these metal ions on the folded fraction of the parallel G-quadruplexes, and UV thermal melting experiments allowed to assess the relative stability of the structures in each cationic condition. Our results show that the RNA quadruplexes are more stable than their DNA counterparts under the same buffer conditions. We have observed that the addition of mainly Na(+), K(+), Rb(+), NH4(+), as well as Sr(2+) and Ba(2+) in water, shifts the equilibrium to the folded quadruplex form, whereby the NRAS sequence responds stronger than TERRA. However, only K(+) and Sr(2+) lead to a significant increase in the stability of the folded structures, which is consistent with their coordination to the O6 atoms from the G-quartet guanosines. Compared to the respective DNA motives, dNRAS and htelo, the RNA sequences are not stabilized by Na(+) ions. Finally, the difference in response between NRAS and TERRA, as well as to the corresponding DNA sequences with respect to different metal ions, could potentially be exploited for selective targeting purposes.
Characterization of the full-length btuB riboswitch from Klebsiella pneumoniae
Palou-Mir, Joana; Musiari, Anastasia; Sigel, Roland K. O.; Barceló-Oliver, Miquel, J. Inorg. Biochem. 2016, 160 , 106-113.
Riboswitches are cis-regulatory RNA elements on the mRNA level that control the expression of the downstream coding region. The interaction of the riboswitch with its specific metabolite, which is related to the function of the controlled gene, induces a structural change of the RNA architecture. Consequently, gene regulation is induced by un/masking of the ribosome binding site (RBS). In the genome of Klebsiella pneumoniae a sequence was identified by bioinformatics and proposed to be a B12 riboswitch regulated by coenzyme B12. Here we study this new coenzyme B12-dependent riboswitch system by in-line probing and ITC. The riboswitch sequence includes the whole expression platform as well as RBS. In-line probing experiments were performed to investigate the structural rearrangement of this 243-nt long RNA sequence while Isothermal Titration Calorimetry (ITC) yielded the thermodynamic parameters of the interaction between the riboswitch and its metabolite. The interaction of coenzyme B12 with the butB riboswitch of K. pneumoniae is an exothermic process with a 1:1 binding stoichiometry and binding affinities of log KA=6.73±0.02 at 15°C and log KA=6.00±0.09 at 30°C.
The X-ray Structures of Six Octameric RNA Duplexes in the Presence of Different Di- and Trivalent Cations
Schaffer, Michelle F.; Peng, Guanya; Spingler, Bernhard; Schnabl, Joachim; Wang, Meitian; Olieric, Vincent; Sigel, Roland K. O., Int. J. Mol. Sci. 2016, 17 , 1-7.
Due to the polyanionic nature of RNA, the principles of charge neutralization and electrostatic condensation require that cations help to overcome the repulsive forces in order for RNA to adopt a three-dimensional structure. A precise structural knowledge of RNA-metal ion interactions is crucial to understand the mechanism of metal ions in the catalytic or regulatory activity of RNA. We solved the crystal structure of an octameric RNA duplex in the presence of the di- and trivalent metal ions Ca(2+), Mn(2+), Co(2+), Cu(2+), Sr(2+), and Tb(3+). The detailed investigation reveals a unique innersphere interaction to uracil and extends the knowledge of the influence of metal ions for conformational changes in RNA structure. Furthermore, we could demonstrate that an accurate localization of the metal ions in the X-ray structures require the consideration of several crystallographic and geometrical parameters as well as the anomalous difference map.
Secondary structure confirmation and localization of Mg2+ ions in the mammalian CPEB3 ribozyme
Skilandat, Miriam; Rowinska-Zyrek, Magdalena; Sigel, Roland K. O., RNA 2016, 22 , 750-763.
Most of today’s knowledge of the CPEB3 ribozyme, one of the few small self-cleaving ribozymes known to occur in humans, is based on comparative studies with the hepatitis delta virus (HDV) ribozyme, which is highly similar in cleavage mechanism and probably also in structure. Here we present detailed NMR studies of the CPEB3 ribozyme in order to verify the formation of the predicted nested double pseudoknot in solution. In particular, the influence of Mg(2+), the ribozyme’s crucial cofactor, on the CPEB3 structure is investigated. NMR titrations, Tb(3+)-induced cleavage, as well as stoichiometry determination by hydroxyquinoline sulfonic acid fluorescence and equilibrium dialysis, are used to evaluate the number, location, and binding mode of Mg(2+)ions. Up to eight Mg(2+)ions interact site-specifically with the ribozyme, four of which are bound with high affinity. The global fold of the CPEB3 ribozyme, encompassing 80{\%}-90{\%} of the predicted base pairs, is formed in the presence of monovalent ions alone. Low millimolar concentrations of Mg(2+)promote a more compact fold and lead to the formation of additional structures in the core of the ribozyme, which contains the inner small pseudoknot and the active site. Several Mg(2+)binding sites, which are important for the functional fold, appear to be located in corresponding locations in the HDV and CPEB3 ribozyme, demonstrating the particular relevance of Mg(2+)for the nested double pseudoknot structure.
An atomistic view on carbocyanine photophysics in the realm of RNA
Steffen, Fabio D.; Sigel, Roland K. O.; Börner, Richard, Phys. Chem. Chem. Phys. 2016, 18 , 29045-29055.
Carbocyanine dyes have a long-standing tradition in fluorescence imaging and spectroscopy, due to their photostability and large spectral separation between individual dye species. Herein, we explore the versatility of cyanine dyes to probe the dynamics of nucleic acids and we report on the interrelation of fluorophores, RNA, and metal ions, namely K(+) and Mg(2+). Photophysical parameters including the fluorescence lifetime, quantum yield and dynamic anisotropy are monitored as a function of the nucleic acid composition, conformation, and metal ion abundance. Occasional excursions to a non-fluorescent cis-state hint at the remarkable sensitivity of carbocyanines to their local environment. Comparison of time-correlated single photon experiments with all-atom molecular dynamics simulations demonstrate that the propensity of photoisomerization is dictated by sterical constraints imposed on the fluorophore. Structural features in the vicinity of the dye play a crucial role in RNA recognition and have far-reaching implications on the mobility of the fluorescent probe. An atomic level description of the mutual interactions will ultimately benefit the quantitative interpretation of single-molecule FRET measurements on large RNA systems.
2015
Explicit analytic equations for multimolecular thermal melting curves
Böttcher, Albrecht; Kowerko, Danny; Sigel, Roland K. O., Biophys. Chem. 2015, 202 , 32-39.
The analysis of thermal melting curves requires the knowledge of equations for the temperature dependence of the relative fraction of folded and unfolded components. To implement these equations as standard tools for curve fitting, they should be as explicit as possible. From the van’t Hoff formalism it is known that the equilibrium constant and hence the folded fraction is a function of the absolute temperature, the van’t Hoff transition enthalpy, and the melting temperature. The work presented here is devoted to the mathematically self-contained derivation and the listing of explicit equations for the folded fraction as a function of the thermodynamic parameters in the case of arbitrary molecularities. Part of the results are known, others are new. It is in particular shown for the first time that the folded fraction is the composition of a universal function which depends solely on the molecularity and a dimensionless function which is governed by the concrete thermodynamic regime but is independent of the molecularity. The results will prove useful for extracting the thermodynamic parameters from experimental data on the basis of regression analysis. As supporting information, open-source Matlab scripts for the computer implementation of the equations are provided.
Mimicking the in vivo Environment – The Effect of Crowding on RNA and Biomacromolecular Folding and Activity
Fiorini, Erica; Börner, Richard; Sigel, Roland K. O., CHIMIA 2015, 69 , 207-212.
In vitro studies on macromolecules, like proteins and nucleic acids, are mostly carried out in highly diluted systems where the molecules are studied under artificial conditions. These experimental conditions are optimized for both the system under investigation and the technique used. However, these conditions often do not reflect the in vivo situation and are therefore inappropriate for a reliable prediction of the native behavior of the molecules and their interactions under in vivo conditions. The intracellular environment is packed with cosolutes (macromolecules, metabolites, etc.) that create ’macromolecular crowding’. The addition of natural or synthetic macromolecules to the sample solution enables crowding to be mimicked. In this surrounding most of the studied biomolecules show a more compact structure, an increased activity, and a decrease of salt requirement for structure formation and function. Herein, we refer to a collection of examples for proteins and nucleic acids and their interactions in crowding environments and present in detail the effect of cosolutes on RNA folding and activity using a group II intron ribozyme as an example.
Cation-induced kinetic heterogeneity of the intron-exon recognition in single group II introns
Kowerko, Danny; König, Sebastian L. B.; Skilandat, Miriam; Kruschel, Daniela; Hadzic, Mélodie C. A. S.; Cardo, Lucia; Sigel, Roland K. O., Proc. Natl. Acad. Sci. U. S. A. 2015, 112 , 3403-3408.
RNA is commonly believed to undergo a number of sequential folding steps before reaching its functional fold, i.e., the global minimum in the free energy landscape. However, there is accumulating evidence that several functional conformations are often in coexistence, corresponding to multiple (local) minima in the folding landscape. Here we use the 5’-exon-intron recognition duplex of a self-splicing ribozyme as a model system to study the influence of Mg(2+) and Ca(2+) on RNA tertiary structure formation. Bulk and single-molecule spectroscopy reveal that near-physiological M(2+) concentrations strongly promote interstrand association. Moreover, the presence of M(2+) leads to pronounced kinetic heterogeneity, suggesting the coexistence of multiple docked and undocked RNA conformations. Heterogeneity is found to decrease at saturating M(2+) concentrations. Using NMR, we locate specific Mg(2+) binding pockets and quantify their affinity toward Mg(2+). Mg(2+) pulse experiments show that M(2+) exchange occurs on the timescale of seconds. This unprecedented combination of NMR and single-molecule Förster resonance energy transfer demonstrates for the first time to our knowledge that a rugged free energy landscape coincides with incomplete occupation of specific M(2+) binding sites at near-physiological M(2+) concentrations. Unconventional kinetics in nucleic acid folding frequently encountered in single-molecule experiments are therefore likely to originate from a spectrum of conformations that differ in the occupation of M(2+) binding sites.
Protonation-Dependent Base Flipping at Neutral pH in the Catalytic Triad of a Self-Splicing Bacterial Group II Intron
Pechlaner, Maria; Donghi, Daniela; Zelenay, Veronika; Sigel, Roland K. O., Angew. Chem. Int. Ed. 2015, 54 , 9687-9690.
NMR spectroscopy has revealed pH-dependent structural changes in the highly conserved catalytic domain 5 of a bacterial group II intron. Two adenines with pK(a) values close to neutral pH were identified in the catalytic triad and the bulge. Protonation of the adenine opposite to the catalytic triad is stabilized within a G(syn)-AH(+) (anti) base pair. The pH-dependent anti-to-syn flipping of this G in the catalytic triad modulates the known interaction with the linker region between domains 2 and 3 (J23) and simultaneously the binding of the catalytic Mg(2+) ion to its backbone. Hence, this here identified shifted pK(a) value controls the conformational change between the two steps of splicing.
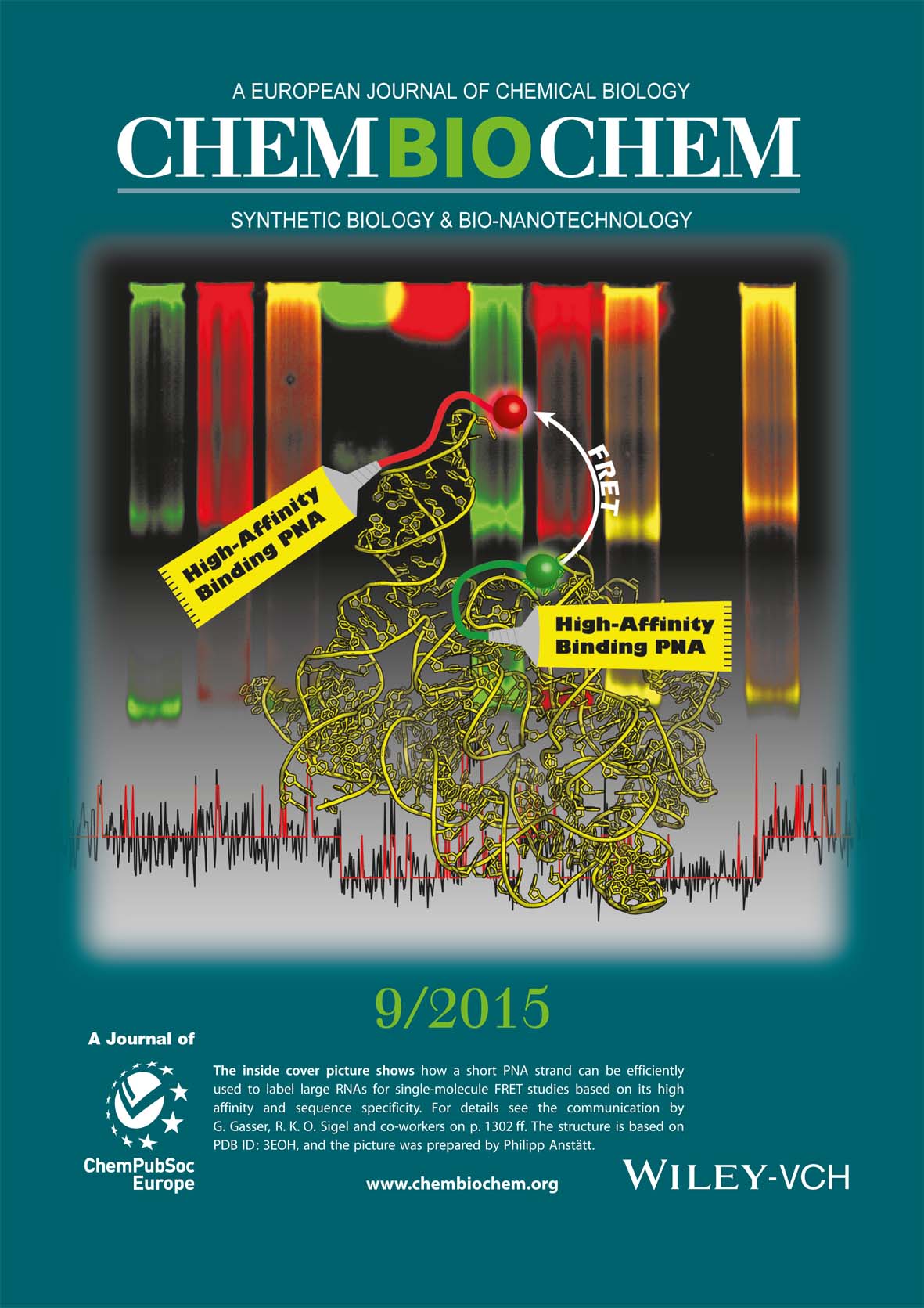
Strategy for Internal Labeling of Large RNAs with Minimal Perturbation by Using Fluorescent PNA
Schmitz, Anita G.; Zelger-Paulus, Susann; Gasser, Gilles; Sigel, Roland K. O., ChemBioChem 2015, 16 , 1302-1306.
Fluorescence techniques for the investigation of biomolecules and their folding pathways require an efficient labeling strategy. A common method to internally label large RNAs involves the introduction of long loops for hybridization of fluorophore-carrying DNA strands. Such loops often disturb the structure, and thus the functionality, of the RNA. Here we show, in a proof of concept study with a >600 nucleotide group II intron ribozyme, that the usage of the nucleic acid analogue peptide nucleic acid (PNA) is more efficient in several aspects, minimizing the required structural modifications of the RNA. We demonstrate by various methods, including smFRET, that much smaller concentrations and shorter PNAs can be applied, compared to DNA, for rapid and specific internal RNA labeling. The folding pathway and catalytic activity of this large ribozyme is only minimally affected by the PNA, but the background signal is significantly reduced.
2014
Mg(2+)-induced conformational changes in the btuB riboswitch from E. coli
Choudhary, Pallavi K.; Sigel, Roland K. O., RNA 2014, 20 , 36-45.
Mg(2+) has been shown to modulate the function of riboswitches by facilitating the ligand-riboswitch interactions. The btuB riboswitch from Escherichia coli undergoes a conformational change upon binding to its ligand, coenzyme B12 (adenosyl-cobalamine, AdoCbl), and down-regulates the expression of the B12 transporter protein BtuB in order to control the cellular levels of AdoCbl. Here, we discuss the structural folding attained by the btuB riboswitch from E. coli in response to Mg(2+) and how it affects the ligand binding competent conformation of the RNA. The btuB riboswitch notably adopts different conformational states depending upon the concentration of Mg(2+). With the help of in-line probing, we show the existence of at least two specific conformations, one being achieved in the complete absence of Mg(2+) (or low Mg(2+) concentration) and the other appearing above ∼0.5 mM Mg(2+). Distinct regions of the riboswitch exhibit different dissociation constants toward Mg(2+), indicating a stepwise folding of the btuB RNA. Increasing the Mg(2+) concentration drives the transition from one conformation toward the other. The conformational state existing above 0.5 mM Mg(2+) defines the binding competent conformation of the btuB riboswitch which can productively interact with the ligand, coenzyme B12, and switch the RNA conformation. Moreover, raising the Mg(2+) concentration enhances the ratio of switched RNA in the presence of AdoCbl. The lack of a AdoCbl-induced conformational switch experienced by the btuB riboswitch in the absence of Mg(2+) indicates a crucial role played by Mg(2+) for defining an active conformation of the riboswitch.
Monitoring global structural changes and specific metal-ion-binding sites in RNA by in-line probing and Tb(III) cleavage
Choudhary, Pallavi K.; Gallo, Sofia; Sigel, Roland K. O., Methods Mol. Biol. 2014, 1086 , 143-158.
In this chapter we describe the use of two methods, in-line probing as well as terbium(III) cleavage. Both methods can be applied to RNAs of any size, structure, and function. Aside from revealing directly metal ion-binding sites these techniques also provide structural information for longer RNA sequences that are out of range to be analyzed with other techniques such as NMR. The cleavage pattern derived from in-line probing experiments reflects local and overall conformational changes in RNA upon the addition of metal ions, metal complexes, or other ligands. On the other side, terbium(III) cleavage experiments are applied to locate specific metal ion-binding sites in RNA molecules.
Synthesis, structure, physico-chemical and biological properties of metal(II) complexes with 5-amino-8-methyl-4H-benzopyran-4-one
Grazul, Magdalena; Sigel, Roland K. O.; Maake, Caroline; Besic-Gyenge, Emina; Lorenz, Ingo-Peter; Mayer, Peter; Czyz, Malgorzata; Budzisz, Elzbieta, Polyhedron 2014, 67 , 136-144.
Two new complexes of a previously obtained chromone derivative, 5-amino-8-methyl-4H-benzopyran-4-one (ligand 1), with the Co(II) (4) and Pd(II) (5) ions were synthesized and characterized by elemental analysis, IR and mass spectroscopy. The previously characterized Cu(II) complexes (2 and 3) and the newly obtained complexes of ligand 1 with Pd(II) and Co(II) have been tested as potential anticancer compounds. The molecular structure of 5 has been determined by X-ray crystallography. Biological properties, such as cytotoxicity against several cancer cell lines, the influence of the obtained compounds on the DNA structure by CD spectroscopy and agarose gel electrophoresis, have been evaluated. Also some physico-chemical properties, such as lipophilicity and stability in the aqueous solution, were tested.
Synthesis, physico-chemical properties and biological analysis of newly obtained copper(II) complexes with pyrazole derivatives
Grazul, Magdalena; Besic-Gyenge, Emina; Maake, Caroline; Ciolkowski, Michal; Czyz, Malgorzata; Sigel, Roland K. O.; Budzisz, Elzbieta, J. Inorg. Biochem. 2014, 135 , 68-76.
Three new copper(II) complexes containing two different pyrazole bound ligands (1, 2) have been synthesized and characterized by IR, LSI-MS (liquid secondary ion mass spectrometry) and elemental analysis. (1)H NMR spectra of the organic ligands have been recorded. We describe the influence of these complexes on particular cancer cell lines and DNA structure by MTT-assay [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide], APA (acid phosphatase activity)-assay or CD-spectroscopy and agarose gel electrophoresis methods, together with their physico-chemical properties such as lipophilicity and stability in aqueous solution. The cytotoxic effect on HUVEC (endothelial cells) for the most active complex 4 has been also investigated. Moreover, the ability of these complexes to induce apoptosis in cancer cells has been assessed by using fluorescence microscopy. Our results indicate that dichloridobis{1-[amino(thioxo)methyl]-5-hydroxy-3-phenyl-1H-pyrazole-κN2}copper(II) is the most potent complex among the tested complexes.
NMR structure of the 5’ splice site in the group IIB intron Sc.ai5γ‐conformational requirements for exon-intron recognition
Kruschel, Daniela; Skilandat, Miriam; Sigel, Roland K. O., RNA 2014, 20 , 295-307.
A crucial step of the self-splicing reaction of group II intron ribozymes is the recognition of the 5’ exon by the intron. This recognition is achieved by two regions in domain 1 of the intron, the exon-binding sites EBS1 and EBS2 forming base pairs with the intron-binding sites IBS1 and IBS2 located at the end of the 5’ exon. The complementarity of the EBS1•IBS1 contact is most important for ensuring site-specific cleavage of the phosphodiester bond between the 5’ exon and the intron. Here, we present the NMR solution structures of the d3’ hairpin including EBS1 free in solution and bound to the IBS1 7-mer. In the unbound state, EBS1 is part of a flexible 11-nucleotide (nt) loop. Binding of IBS1 restructures and freezes the entire loop region. Mg(2+) ions are bound near the termini of the EBS1•IBS1 helix, stabilizing the interaction. Formation of the 7-bp EBS1•IBS1 helix within a loop of only 11 nt forces the loop backbone to form a sharp turn opposite of the splice site, thereby presenting the scissile phosphate in a position that is structurally unique.
Photo-induced uncaging of a specific Re( i ) organometallic complex in living cells
Leonidova, Anna; Pierroz, Vanessa; Rubbiani, Riccardo; Lan, Yanjun; Schmitz, Anita G.; Kaech, Andres; Sigel, Roland K. O.; Ferrari, Stefano; Gasser, Gilles, Chem. Sci. 2014, 5 , 4044.
In the last decades, a large number of organometallic complexes have shown promising anti-proliferative activity towards different cancer cell lines. However, these compounds generally had low cellular uptake and low selectivity towards cancer cells over healthy cells. The use of external triggers (e.g. light, ultra-sound, temperature, etc.) to modify the cytotoxic effect of a prodrug and the coupling of a targeting vector (e.g. peptides, antibodies, etc.) to a drug were found to be very successful techniques to tackle these drawbacks. Here, we envisioned combining these two methods, namely an external trigger (i.e. light activation) and a targeting vector, in an organometallic compound. More specifically, a Re(I) tricarbonyl N,N-bis(quinolinoyl) complex (Re-NH2) was derivatised with a photo-labile protecting group (PLPG) to cage Re-NH2 by formation of Re-PLPG. For organelle/cellular specificity, Re-PLPG was then further coupled to a nuclear localization sequence (NLS) or a bombesin peptide derivative to give Re-PLPG-NLS or Re-PLPG-Bombesin, respectively. Photolysis experiments in PBS buffer (pH 7.4) demonstrated that Re-NH2 was completely photo-released from Re-PLPG-NLS and Re-PLPG-Bombesin using a very low irradiation dose (1.2 J cm−2). To the best of our knowledge, these are the first two examples of the selective photo-release of an intact organometallic compound from a bioconjugate. Of high interest, both derivatives showed toxicity comparable to that of cisplatin towards cervical cancer cells (HeLa) upon light irradiation, although the phototoxic index (PTI) varied greatly with the targeting peptide. The cell death mechanism of Re-PLPG-NLS was explored using different techniques, including fluorescence microscopy, ICP-MS, gel electrophoresis, flow cytometry and transmission electron microscopy (TEM). It could be demonstrated that HeLa cells treated with Re-PLPG-NLS in the dark and upon irradiation showed severe cell stress (nucleolar segregation, pyknosis and vacuolation). The data obtained from an Annexin V/propidium iodide (PI) assay indicated that, after an early apoptotic stage, the onset induced by Re-PLPG-NLS led to cell death, with features ascribable to late apoptosis and necrosis, which were more marked for the treatment involving irradiation.
The AdoCbl-riboswitch interaction investigated by in-line probing and surface plasmon resonance spectroscopy (SPR)
Schaffer, Michelle F.; Choudhary, Pallavi K.; Sigel, Roland K. O., Methods Enzymol. 2014, 549 , 467-488.
The unique feature of riboswitches is to control selected gene expression by specific recognition of a cognate ligand. AdoCbl (adenosyl cobalamin, coenzyme B12) riboswitches regulate the expression of enzymes and transporters involved in bacterial AdoCbl biosynthesis or uptake on a transcriptional and/or translational level. The analysis of ligand recognition and the induced conformational changes requires a detailed knowledge of the kinetics and thermodynamics of ligand binding and of the secondary structure rearrangements. This chapter describes the investigation of coenzyme B12 binding to the btuB riboswitch from Escherichia coli by in-line probing assays and surface plasmon resonance spectroscopy. The experimental conditions, requirements, and performance of both methods are presented together with the evaluation of the experimental data to determine the associated conformational changes of the RNA and the kinetic and thermodynamic parameters of ligand binding. Owing to the light sensitivity of the cobalt(I)’carbon bond, these methods were specifically modified to ensure the chemical integrity of AdoCbl.
The role of Mg(II) in DNA cleavage site recognition in group II intron ribozymes: Solution structure and metal ion binding sites of the RNA-DNA complex
Skilandat, Miriam; Sigel, Roland K. O., J. Biol. Chem. 2014, 289 , 20650-20663.
Group II intron ribozymes catalyze the cleavage of (and their reinsertion into) DNA and RNA targets using a Mg2(+)-dependent reaction. The target is cleaved 3’ to the last nucleotide of intron binding site 1 (IBS1), one of three regions that form base pairs with the intron’s exon binding sites (EBS1 to -3).We solved the NMR solution structure of the d3’ hairpin of the Sc.ai5γ intron containing EBS1 in its 11-nucleotide loop in complex with the dIBS1 DNA 7-mer and compare it with the analogous RNA-RNA contact. The EBS1-dIBS1 helix is slightly flexible and non-symmetric. NMR data reveal two major groove binding sites for divalent metal ions at the EBS1-dIBS1 helix, and surface plasmon resonance experiments show that low concentrations of Mg2(+) considerably enhance the affinity of dIBS1 for EBS1. Our results indicate that identification of both RNA and DNA IBS1 targets, presentation of the scissile bond, and stabilization of the structure by metal ions are governed by the overall structure of EBS1-dIBS1 and the surrounding loop nucleotides but are irrespective of different EBS1-(d)IBS1 geometries and interstrand affinities.
Solution structure and metal ion binding sites of the human CPEB3 ribozyme’s P4 domain
Skilandat, Miriam; Rowinska-Zyrek, Magdalena; Sigel, Roland K. O., J. Biol. Inorg. Chem. 2014, 19 , 903-912.
Three ribozymes are known to occur in humans, the CPEB3 ribozyme, the CoTC ribozyme, and the hammerhead ribozyme. Here, we present the NMR solution structure of a well-conserved motif within the CPEB3 ribozyme, the P4 domain. In addition, we discuss the binding sites and impact of Mg(2+) and [Co(NH3)6](3+), a spectroscopic probe for [Mg(H2O)6](2+), on the structure. The well-defined P4 region is a hairpin closed with a UGGU tetraloop that shows a distinct electrostatic surface potential and a characteristic, strongly curved backbone trajectory. The P4 hairpin contains two specific Mg(2+) binding sites: one outer-sphere binding site close to the proposed CPEB3 ribozyme active site with potential relevance for maintaining a compact fold of the ribozyme core, and one inner-sphere binding site, probably stabilizing the tetraloop structure. The structure of the tetraloop resembles an RNase III recognition structure, as previously described for an AGUU tetraloop. The detailed knowledge of the P4 domain and its metal ion binding preferences thus brings us closer to understanding the importance of Mg(2+) binding for the CPEB3 ribozyme’s fold and function in the cell.
Structure determination of noncanonical RNA motifs guided by ¹H NMR chemical shifts
Sripakdeevong, Parin; Cevec, Mirko; Chang, Andrew T.; Erat, Michèle C.; Ziegeler, Melanie; Zhao, Qin; Fox, George E.; Gao, Xiaolian; Kennedy, Scott D.; Kierzek, Ryszard; Nikonowicz, Edward P.; Schwalbe, Harald; Sigel, Roland K. O.; Turner, Douglas H.; Das, Rhiju, Nat. Methods 2014, 11 , 413-416.
Structured noncoding RNAs underlie fundamental cellular processes, but determining their three-dimensional structures remains challenging. We demonstrate that integrating ¹H NMR chemical shift data with Rosetta de novo modeling can be used to consistently determine high-resolution RNA structures. On a benchmark set of 23 noncanonical RNA motifs, including 11 ’blind’ targets, chemical-shift Rosetta for RNA (CS-Rosetta-RNA) recovered experimental structures with high accuracy (0.6-2.0 Å all-heavy-atom r.m.s. deviation) in 18 cases.
2013
Diversity of cobalamin riboswitches in the corrinoid-producing organohalide respirer Desulfitobacterium hafniense
Choudhary, Pallavi K.; Duret, Aurélie; Rohrbach-Brandt, Emmanuelle; Holliger, Christof; Sigel, Roland K. O.; Maillard, Julien, J. Bacteriol. 2013, 195 , 5186-5195.
The strategic adaptation of prokaryotes in polluted niches involves the efficient regulation of their metabolism. The obligate anaerobe and metabolically versatile Desulfitobacterium hafniense reductively dechlorinates halogenated organic compounds (so-called organohalides). Some D. hafniense strains carry out organohalide respiration (OHR), a process which requires the use of corrinoid as a cofactor in reductive dehalogenases, the key enzymes in OHR. We report here the diversity of the cobalamin riboswitches that possibly regulate the corrinoid metabolism for D. hafniense. The analysis of available D. hafniense genomes indicates the presence of 18 cobalamin riboswitches located upstream of genes whose products are mainly involved in corrinoid biosynthesis and transport. To obtain insight into their function, the secondary structures of three of these RNA elements were predicted by Mfold, as well as analyzed by in-line probing. These RNA elements both display diversity in their structural elements and exhibit various affinities toward adenosylcobalamin that possibly relates to their role in the regulation of corrinoid metabolism. Furthermore, adenosylcobalamin-induced in vivo repression of RNA synthesis of the downstream located genes indicates that the corrinoid transporters and biosynthetic enzymes in D. hafniense strain TCE1 are regulated at the transcriptional level. Taken together, the riboswitch-mediated regulation of the complex corrinoid metabolism in D. hafniense could be of crucial significance in environments polluted with organohalides both to monitor their intracellular corrinoid level and to coexist with corrinoid-auxotroph OHR bacteria.
Intrinsic acid-base properties of a hexa-2’-deoxynucleoside pentaphosphate, d(ApGpGpCpCpT): neighboring effects and isomeric equilibria
Domínguez-Martín, Alicia; Johannsen, Silke; Sigel, Astrid; Operschall, Bert P.; Song, Bin; Sigel, Helmut; Okruszek, Andrzej; González-Pérez, Josefa María; Niclós-Gutiérrez, Juan; Sigel, Roland K. O., Chem. Eur. J. 2013, 19 , 8163-8181.
The intrinsic acid-base properties of the hexa-2’-deoxynucleoside pentaphosphate, d(ApGpGpCpCpT) [=(A1∙G2∙G3∙C4∙C5∙T6)=(HNPP)⁵⁻] have been determined by ¹H NMR shift experiments. The pKa values of the individual sites of the adenosine (A), guanosine (G), cytidine (C), and thymidine (T) residues were measured in water under single-strand conditions (i.e., 10{\%} D₂O, 47 °C, I=0.1 M, NaClO₄). These results quantify the release of H⁺ from the two (N7)H⁺ (G∙G), the two (N3)H⁺ (C∙C), and the (N1)H⁺ (A) units, as well as from the two (N1)H (G∙G) and the (N3)H (T) sites. Based on measurements with 2’-deoxynucleosides at 25 °C and 47 °C, they were transferred to pKa values valid in water at 25 °C and I=0.1 M. Intramolecular stacks between the nucleobases A1 and G2 as well as most likely also between G2 and G3 are formed. For HNPP three pKa clusters occur, that is those encompassing the pKa values of 2.44, 2.97, and 3.71 of G2(N7)H⁺, G3(N7)H⁺, and A1(N1)H⁺, respectively, with overlapping buffer regions. The tautomer populations were estimated, giving for the release of a single proton from five-fold protonated H₅(HNPP)(±) , the tautomers (G2)N7, (G3)N7, and (A1)N1 with formation degrees of about 74, 22, and 4{\%}, respectively. Tautomer distributions reveal pathways for proton-donating as well as for proton-accepting reactions both being expected to be fast and to occur practically at no \{\textquotedbl}cost\{\textquotedbl}. The eight pKa values for H₅(HNPP)(±) are compared with data for nucleosides and nucleotides, revealing that the nucleoside residues are in part affected very differently by their neighbors. In addition, the intrinsic acidity constants for the RNA derivative r(A1∙G2∙G3∙C4∙C5∙U6), where U=uridine, were calculated. Finally, the effect of metal ions on the pKa values of nucleobase sites is briefly discussed because in this way deprotonation reactions can easily be shifted to the physiological pH range.
The structural stabilization of the κ three-way junction by Mg(II) represents the first step in the folding of a group II intron
Donghi, Daniela; Pechlaner, Maria; Finazzo, Cinzia; Knobloch, Bernd; Sigel, Roland K. O., Nucleic Acids Res. 2013, 41 , 2489-2504.
Folding of group II introns is characterized by a first slow compaction of domain 1 (D1) followed by the rapid docking of other domains to this scaffold. D1 compaction initiates in a small subregion encompassing the κ and ζ elements. These two tertiary elements are also the major interaction sites with domain 5 to form the catalytic core. Here, we provide the first characterization of the structure adopted at an early folding step and show that the folding control element can be narrowed down to the three-way junction with the κ motif. In our nuclear magnetic resonance studies of this substructure derived from the yeast mitochondrial group II intron Sc.ai5γ, we show that a high affinity Mg(II) ion stabilizes the κ element and enables coaxial stacking between helices d’ and d”, favoring a rigid duplex across the three-way junction. The κ-element folds into a stable GAAA-tetraloop motif and engages in A-minor interactions with helix d’. The addition of cobalt(III)hexammine reveals three distinct binding sites. The Mg(II)-promoted structural rearrangement and rigidification of the D1 core can be identified as the first micro-step of D1 folding.
Helicase-mediated changes in RNA structure at the single-molecule level
König, Sebastian L. B.; Liyanage, Pramodha S.; Sigel, Roland K. O.; Rueda, David, RNA Biol. 2013, 10 , 133-148.
RNA helicases are a diverse group of RNA-dependent ATPases known to play a large number of biological roles inside the cell, such as RNA unwinding, remodeling, export and degradation. Understanding how helicases mediate changes in RNA structure is therefore of fundamental interest. The advent of single-molecule spectroscopic techniques has unveiled with unprecedented detail the interplay of RNA helicases with their substrates. In this review, we describe the characterization of helicase-RNA interactions by single-molecule approaches. State-of-the-art techniques are presented, followed by a discussion of recent advancements in this exciting field.
Kinetic Subpopulations Detected by Single-molecule Spectroscopy: Fundamental Property of Functional Nucleic Acids or Experimental Artefact?
König, Sebastian L. B.; Kowerko, Danny; Sigel, Roland K. O., CHIMIA 2013, 67 , 240-243.
Single-molecule spectroscopy allows the direct observation of conformational dynamics in individual biomolecules. Here, we describe how single-molecule Förster resonance energy transfer (smFRET) reveals heterogeneous kinetics in the EBS1*/IBS1* interaction, two RNA sequences that play an important role in group II intron mediated self-cleavage. Further examples of dynamic heterogeneity in functional nucleic acids are provided and the possible origins of this phenomenon are discussed.
Distance-dependent duplex DNA destabilization proximal to G-quadruplex/i-motif sequences
König, Sebastian L. B.; Huppert, Julian L.; Sigel, Roland K. O.; Evans, Amanda C., Nucleic Acids Res. 2013, 41 , 7453-7461.
G-quadruplexes and i-motifs are complementary examples of non-canonical nucleic acid substructure conformations. G-quadruplex thermodynamic stability has been extensively studied for a variety of base sequences, but the degree of duplex destabilization that adjacent quadruplex structure formation can cause has yet to be fully addressed. Stable in vivo formation of these alternative nucleic acid structures is likely to be highly dependent on whether sufficient spacing exists between neighbouring duplex- and quadruplex-/i-motif-forming regions to accommodate quadruplexes or i-motifs without disrupting duplex stability. Prediction of putative G-quadruplex-forming regions is likely to be assisted by further understanding of what distance (number of base pairs) is required for duplexes to remain stable as quadruplexes or i-motifs form. Using oligonucleotide constructs derived from precedented G-quadruplexes and i-motif-forming bcl-2 P1 promoter region, initial biophysical stability studies indicate that the formation of G-quadruplex and i-motif conformations do destabilize proximal duplex regions. The undermining effect that quadruplex formation can have on duplex stability is mitigated with increased distance from the duplex region: a spacing of five base pairs or more is sufficient to maintain duplex stability proximal to predicted quadruplex/i-motif-forming regions.
BOBA FRET: Bootstrap-based analysis of single-molecule FRET data
König, Sebastian L. B.; Hadzic, Mélodie C. A. S.; Fiorini, Erica; Börner, Richard; Kowerko, Danny; Blanckenhorn, Wolf U.; Sigel, Roland K. O., PLoS One 2013, 8 , e84157.
Time-binned single-molecule Förster resonance energy transfer (smFRET) experiments with surface-tethered nucleic acids or proteins permit to follow folding and catalysis of single molecules in real-time. Due to the intrinsically low signal-to-noise ratio (SNR) in smFRET time traces, research over the past years has focused on the development of new methods to extract discrete states (conformations) from noisy data. However, limited observation time typically leads to pronounced cross-sample variability, i.e., single molecules display differences in the relative population of states and the corresponding conversion rates. Quantification of cross-sample variability is necessary to perform statistical testing in order to assess whether changes observed in response to an experimental parameter (metal ion concentration, the presence of a ligand, etc.) are significant. However, such hypothesis testing has been disregarded to date, precluding robust biological interpretation. Here, we address this problem by a bootstrap-based approach to estimate the experimental variability. Simulated time traces are presented to assess the robustness of the algorithm in conjunction with approaches commonly used in thermodynamic and kinetic analysis of time-binned smFRET data. Furthermore, a pair of functionally important sequences derived from the self-cleaving group II intron Sc.ai5γ (d3’EBS1/IBS1) is used as a model system. Through statistical hypothesis testing, divalent metal ions are shown to have a statistically significant effect on both thermodynamic and kinetic aspects of their interaction. The Matlab source code used for analysis (bootstrap-based analysis of smFRET data, BOBA FRET), as well as a graphical user interface, is available via http://www.aci.uzh.ch/rna/.
A QM/MM refinement of an experimental DNA structure with metal-mediated base pairs
Kumbhar, Sadhana; Johannsen, Silke; Sigel, Roland K. O.; Waller, Mark P.; Müller, Jens, J. Inorg. Biochem. 2013, 127 , 203-210.
A series of hybrid quantum mechanical/molecular mechanical (QM/MM) calculations was performed on models of a DNA duplex with artificial silver(I)-mediated imidazole base pairs. The optimized structures were compared to the original experimental NMR structure (Nat. Chem. 2 (2010) 229-234). The metal⋯metal distances are significantly shorter (~0.5Å) in the QM/MM model than in the original NMR structure. As a result, argentophilic interactions are feasible between the silver(I) ions of neighboring metal-mediated base pairs. Using the computationally determined metal⋯metal distances, a re-refined NMR solution structure of the DNA duplex was obtained. In this new NMR structure, all experimental constraints remain fulfilled. The new NMR structure shows less deviation from the regular B-type conformation than the original one. This investigation shows that the application of QM/MM models to generate additional constraints to be used during NMR structural refinements represents an elegant approach to obtaining high-resolution NMR structures.
Structure and conformational dynamics of the domain 5 RNA hairpin of a bacterial group II intron revealed by solution nuclear magnetic resonance and molecular dynamics simulations
Pechlaner, Maria; Sigel, Roland K. O.; van Gunsteren, Wilfred F.; Dolenc, Jožica, Biochemistry 2013, 52 , 7099-7113.
Nuclear magnetic resonance (NMR) nuclear Overhauser enhancement (NOE) data obtained for a 35-nucleotide RNA segment of a bacterial group II intron indicate a helical hairpin structure in which three parts, a terminal pentaloop, a bulge, and a G-A mismatch, display no Watson-Crick base pairing. The 668 NOE upper distance bounds for atom pairs are insufficient to uniquely determine the conformation of these segments. Therefore, molecular dynamics simulations including time-averaged distance restraints have been used to obtain a conformational ensemble compatible with the observed NMR data. The ensemble shows alternating hydrogen bonding patterns for the mentioned segments. In particular, in the pentaloop and in the bulge, the hydrogen bonding networks correspond to distinct conformational clusters that could not be captured by using conventional single-structure refinement techniques. This implies that, to obtain a realistic picture of the conformational ensemble of such flexible biomolecules, it is necessary to properly account for the conformational variability in the structure refinement of RNA fragments.
Binding of a designed anti-cancer drug to the central cavity of an RNA three-way junction
Phongtongpasuk, Siriporn; Zelger-Paulus, Susann; Schnabl, Joachim; Sigel, Roland K. O.; Spingler, Bernhard; Hannon, Michael J.; Freisinger, Eva, Angew. Chem. Int. Ed. 2013, 52 , 11513-11516.
Getting to the heart of it: Co-crystallization of an RNA three-way junction with a cylindrical di-iron(II)-based anti-cancer drug (green) results in π-stacking interactions between the cylinder and the central base pairs of the RNA structure. The shape, size, and cationic nature of the cylinder were found to be responsible for this perfect fit. Native gel electrophoresis studies confirmed stabilization of the RNA three-way junction by the iron(II) cylinder.
Hexaamminecobalt(III) - Probing Metal Ion Binding Sites in Nucleic Acids by NMR Spectroscopy
Rowinska-Zyrek, Magdalena; Skilandat, Miriam; Sigel, Roland K. O., Z. Anorg. Allg. Chem. 2013, 639 , 1313-1320.
Hexaamminecobalt(III), an octahedral, inert metal ion complex, has lately gained increasing attention of structural biologists and bioinorganic chemists due to its use in structure determination of nucleic acids. This complex mimics outer-sphere binding events of the physiologically relevant magnesium(II)hexaaqua ion; hexaamminecobalt(III) often finds usage either in NMR spectroscopy, where cross-peaks between the complex ammines and the nucleic acid protons near the binding site are observed, in X-ray spectroscopy as heavy metal derivative for phasing, or in other techniques. In this review, we discuss the basic hexaamminecobalt(III) binding modes and give an overview on the most recent findings on [Co(NH3)6]Cl3–nucleic acid complexes. The various techniques that are applied in combination with this complex are mentioned and briefly summarized. Special attention is given to the application of [Co(NH3)6]Cl3 in nuclear magnetic resonance spectroscopy of nucleic acids, where it is used to reveal potential outer-sphere magnesium binding sites.
Complex formation of cadmium with sugar residues, nucleobases, phosphates, nucleotides, and nucleic acids
Sigel, Roland K. O.; Skilandat, Miriam; Sigel, Astrid; Operschall, Bert P.; Sigel, Helmut, Met. Ions Life Sci. 2013, 11 , 191-274.
Cadmium(II), commonly classified as a relatively soft metal ion, prefers indeed aromatic-nitrogen sites (e.g., N7 of purines) over oxygen sites (like sugar-hydroxyl groups). However, matters are not that simple, though it is true that the affinity of Cd(2+) towards ribose-hydroxyl groups is very small; yet, a correct orientation brought about by a suitable primary binding site and a reduced solvent polarity, as it is expected to occur in a folded nucleic acid, may facilitate metal ion-hydroxyl group binding very effectively. Cd(2+) prefers the guanine(N7) over the adenine(N7), mainly because of the steric hindrance of the (C6)NH(2) group in the adenine residue. This Cd(2+)-(N7) interaction in a guanine moiety leads to a significant acidification of the (N1)H meaning that the deprotonation reaction occurs now in the physiological pH range. N3 of the cytosine residue, together with the neighboring (C2)O, is also a remarkable Cd(2+) binding site, though replacement of (C2)O by (C2)S enhances the affinity towards Cd(2+) dramatically, giving in addition rise to the deprotonation of the (C4)NH(2) group. The phosphodiester bridge is only a weak binding site but the affinity increases further from the mono- to the di- and the triphosphate. The same also holds for the corresponding nucleotides. Complex stability of the pyrimidine-nucleotides is solely determined by the coordination tendency of the phosphate group(s), whereas in the case of purine-nucleotides macrochelate formation takes place by the interaction of the phosphate-coordinated Cd(2+) with N7. The extents of the formation degrees of these chelates are summarized and the effect of a non-bridging sulfur atom in a thiophosphate group (versus a normal phosphate group) is considered. Mixed ligand complexes containing a nucleotide and a further mono- or bidentate ligand are covered and it is concluded that in these species N7 is released from the coordination sphere of Cd(2+). In the case that the other ligand contains an aromatic residue (e.g., 2,2’-bipyridine or the indole ring of tryptophanate) intramolecular stack formation takes place. With buffers like Tris or Bistris mixed ligand complexes are formed. Cd(2+) coordination to dinucleotides and to dinucleoside monophosphates provides some insights regarding the interaction between Cd(2+) and nucleic acids. Cd(2+) binding to oligonucleotides follows the principles of coordination to its units. The available crystal studies reveal that N7 of purines is the prominent binding site followed by phosphate oxygens and other heteroatoms in nucleic acids. Due to its high thiophilicity, Cd(2+) is regularly used in so-called thiorescue experiments, which lead to the identification of a direct involvement of divalent metal ions in ribozyme catalysis.
2012
Single molecule FRET characterization of large ribozyme folding
Cardo, Lucia; Karunatilaka, Krishanthi S.; Rueda, David; Sigel, Roland K. O., Methods Mol. Biol. 2012, 848 , 227-251.
A procedure to investigate the folding of group II intron by single molecule Fluorescence Resonance Energy Transfer (smFRET) using total internal reflection fluorescence microscopy (TIRFM) is described in this chapter. Using our previous studies on the folding and dynamics of a large ribozyme in the presence of metal ions (i.e., Mg(2+) and Ca(2+)) and/or the DEAD-box protein Mss116 as an example, we here describe step-by-step procedures to perform experiments. smFRET allows the investigation of individual molecules, thus, providing kinetic and mechanistic information hidden in ensemble averaged experiments.
Metal ion-RNA interactions studied via multinuclear NMR
Donghi, Daniela; Sigel, Roland K. O., Methods Mol. Biol. 2012, 848 , 253-273.
Metal ions are indispensable for ribonucleic acids (RNAs) folding and activity. First they act as charge neutralization agents, allowing the RNA molecule to attain the complex active three dimensional structure. Second, metal ions are eventually directly involved in function. Nuclear magnetic resonance (NMR) spectroscopy offers several ways to study the RNA-metal ion interactions at an atomic level. Here, we first focus on special requirements for NMR sample preparation for this kind of experiments: the practical aspects of in vitro transcription and purification of small (<50 nt) RNA fragments are described, as well as the precautions that must be taken into account when a sample for metal ion titration experiments is prepared. Subsequently, we discuss the NMR techniques to accurately locate and characterize metal ion binding sites in a large RNA. For example, (2) J-[(1)H,(15)N]-HSQC (heteronuclear single quantum coherence) experiments are described to qualitatively distinguish between different modes of interaction. Finally, part of the last section is devoted to data analysis; this is how to calculate intrinsic affinity constants.
NMR spectroscopy in bioinorganic chemistry
Donghi, Daniela; Johannsen, Silke; Sigel, Roland K. O.; Freisinger, Eva, CHIMIA 2012, 66 , 791-797.
Multinuclear and multidimensional nuclear magnetic resonance (NMR) spectroscopy is applied in our groups to gain insights into the role of metal ions for the function and structure of large biomolecules. Specifically, NMR is used i) to investigate how metal ions bind to nucleic acids and thereby control the folding and structure of RNAs, ii) to characterize how metal ions are able to stabilize modified nucleic acids to be used as potential nanowires, and iii) to characterize the formation, structure, and role of the diverse metal clusters within plant metallothioneins. In this review we summarize the various NMR experiments applied and the information obtained, demonstrating the important and fascinating part NMR spectroscopy plays in the field of bioinorganic chemistry.
Accurate analysis of Mg2+ binding to RNA: From classical methods to a novel iterative calculation procedure - Review
Erat, Michèle C.; Coles, Jonathan; Finazzo, Cinzia; Knobloch, Bernd; Sigel, Roland K. O., Coord. Chem. Rev. 2012, 256 , 279-288.
Mg2+ acts as a catalytic cofactor in many ribozymes and specifically bound divalent metal ions have been implicated in the stabilization of structural motifs that are essential for RNA folding. The accurate calculation of intrinsic affinity constants of M2+ to specific binding sites in nucleic acids is therefore of high importance. Methods classically applied to determine the affinity constants of metal ions to RNAs are summarized in the first part of this review, e.g. hydrolytic cleavage experiments, equilibrium dialysis, and spectroscopic techniques like EPR and NMR. However, the fact that several binding sites of similar affinities are often present in a single RNA molecule is mostly neglected. The most immediate consequence of several binding sites is that less than the total amount of M2+ is available to bind to a particular binding site at a given total concentration. We have recently introduced a new iterative procedure that tackles this problem and have developed a rapid calculation tool (ISTAR) that is available from the authors. Here, we explain this procedure in detail under different assumptions and illustrate how the intrinsic affinity constants for Mg2+ to a short RNA hairpin, a minimal domain 6 from the group II intron Sc.ai5 gamma, change. We use ISTAR to calculate intrinsic affinities and to validate a particular binding stoichiometry by judging the quality of the fit to the experimental data for a given model. This is important since weak coordination sites exhibiting similar binding affinities, and being thus in direct competition to each other, are a characteristic feature of nucleic acids. With ISTAR these binding affinities can be calculated more accurately within minutes and we can gain a better understanding of these crucial metal ion-nucleic acid interactions. (C) 2011 Published by Elsevier B.V.
Unusually high-affinity Mg(2+) binding at the AU-rich sequence within the antiterminator hairpin of a Mg(2+) riboswitch
Korth, Maximiliane M. T.; Sigel, Roland K. O., Chem. Biodiv. 2012, 9 , 2035-2049.
Mg(2+)-Responsive riboswitches represent a fascinating example of bifunctional RNAs that sense Mg(2+) ions with high selectivity and autonomously regulate the expression of Mg(2+)-transporter proteins. The mechanism of the mgtA riboswitch is scarcely understood, and a detailed structural analysis is called for to study how this RNA can selectively recognize Mg(2+) and respond by switching between two alternative stem loop structures. In this work, we investigated the structure and Mg(2+)-binding properties of the lower part of the antiterminator loop C from the mgtA riboswitch of Yersinia enterocolitica by solution NMR and report a discrete Mg(2+)-binding site embedded in the AU-rich sequence. At the position of Mg(2+) binding, the helical axis exhibits a distinct kink accompanied by a widening of the major groove, which accommodates the Mg(2+)-binding pocket. An unusually large overlap between two adenine residues on the opposite strands suggests that the bending may be sequence-induced by strong stacking interactions, enabling Mg(2+) to bind at this so-far not described metal-ion binding site.
Synthesis and acid-base properties of an imidazole-containing nucleotide analog, 1-(2’-deoxy-β-D-ribofuranosyl)imidazole 5’-monophosphate (dImMP(2-))
Megger, Nicole; Johannsen, Silke; Müller, Jens; Sigel, Roland K. O., Chem. Biodiv. 2012, 9 , 2050-2063.
Deletion of the substituted pyrimidine ring in purine-2’-deoxynucleoside 5’-monophosphates leads to the artificial nucleotide analog dImMP(2-). This analog can be incorporated into DNA to yield, upon addition of Ag(+) ions, a molecular wire. Here, we measured the acidity constants of H(2)(dImMP)(±) having one proton at N(3) and one at the PO(3)(2-) group by potentiometric pH titrations in aqueous solution. The micro acidity constants show that N(3) is somewhat more basic than PO(3)(2-) and, consequently, the (H·dImMP)(-) tautomer with the proton at N(3) dominates to ca. 75{\%}. The calculated micro acidity constants are confirmed by (31)P- and (1)H-NMR chemical shifts. The assembled data allow many quantitative comparisons, e.g., the N(3)-protonated and thus positively charged imidazole residue facilitates deprotonation of the P(O)(2)(OH)(-) group by 0.3 pK units. Information on the intrinsic site basicities also allows predictions about metal-ion binding; e.g., Mg(2+) and Mn(2+) will primarily coordinate to the phosphate group, whereas Ni(2+) and Cu(2+) will preferably bind to N(3). Macrochelate formation for these metal ions is also predicted. The micro acidity constant for N(3)H(+) deprotonation in the (H·dImMP·H)(±) species (pk(a) 6.46) and the M(n+)-binding properties are of relevance for understanding the behavior of dImMP units present in DNA hairpins and metalated duplexes.
Characterization of metal ion-nucleic acid interactions in solution
Pechlaner, Maria; Sigel, Roland K. O., Met. Ions Life Sci. 2012, 10 , 1-42.
Metal ions are inextricably involved with nucleic acids due to their polyanionic nature. In order to understand the structure and function of RNAs and DNAs, one needs to have detailed pictures on the structural, thermodynamic, and kinetic properties of metal ion interactions with these biomacromolecules. In this review we first compile the physicochemical properties of metal ions found and used in combination with nucleic acids in solution. The main part then describes the various methods developed over the past decades to investigate metal ion binding by nucleic acids in solution. This includes for example hydrolytic and radical cleavage experiments, mutational approaches, as well as kinetic isotope effects. In addition, spectroscopic techniques like EPR, lanthanide(III) luminescence, IR and Raman as well as various NMR methods are summarized. Aside from gaining knowledge about the thermodynamic properties on the metal ion-nucleic acid interactions, especially NMR can be used to extract information on the kinetics of ligand exchange rates of the metal ions applied. The final section deals with the influence of anions, buffers, and the solvent permittivity on the binding equilibria between metal ions and nucleic acids. Little is known on some of these aspects, but it is clear that these three factors have a large influence on the interaction between metal ions and nucleic acids.
MINAS‐a database of Metal Ions in Nucleic AcidS
Schnabl, Joachim; Suter, Pascal; Sigel, Roland K. O., Nucleic Acids Res. 2012, 40 , D434-8.
Correctly folded into the respective native 3D structure, RNA and DNA are responsible for uncountable key functions in any viable organism. In order to exert their function, metal ion cofactors are closely involved in folding, structure formation and, e.g. in ribozymes, also the catalytic mechanism. The database MINAS, Metal Ions in Nucleic AcidS (http://www.minas.uzh.ch), compiles the detailed information on innersphere, outersphere and larger coordination environment of >70,000 metal ions of 36 elements found in >2000 structures of nucleic acids contained today in the PDB and NDB. MINAS is updated monthly with new structures and offers a multitude of search functions, e.g. the kind of metal ion, metal-ligand distance, innersphere and outersphere ligands defined by element or functional group, residue, experimental method, as well as PDB entry-related information. The results of each search can be saved individually for later use with so-called miniPDB files containing the respective metal ion together with the coordination environment within a 15 Å radius. MINAS thus offers a unique way to explore the coordination geometries and ligands of metal ions together with the respective binding pockets in nucleic acids.
2011
Probing the metal-ion-binding strength of the hydroxyl group - Reveiw
Al-Sogair, Fawzia M.; Operschall, Bert P.; Sigel, Astrid; Sigel, Helmut; Schnabl, Joachim; Sigel, Roland K. O., Chem. Rev. 2011, 111 , 4964-5003.
There is no abstract for this review.
The change of corrin-amides to carboxylates leads to altered structures of the B12-responding btuB riboswitch
Gallo, Sofia; Mundwiler, Stefan; Alberto, Roger; Sigel, Roland K. O., Chem. Commun. 2011, 47 , 403-405.
By applying four different acid derivatives of vitamin B(12), we demonstrate that the H-bonding pattern and the electrostatic environment provided by each side chain of the corrin ring are crucial for the correct structural rearrangement of the btuB riboswitch of E. coli.
Stability and structure of mixed-ligand metal ion complexes that contain Ni2+, Cu2+, or Zn2+, and Histamine, as well as adenosine 5’-triphosphate (ATP4-) or uridine 5’-triphosphate (UTP(4-): An intricate network of equilibria
Knobloch, Bernd; Mucha, Ariel; Operschall, Bert P.; Sigel, Helmut; Jeżowska-Bojczuk, Małgorzata; Kozłowski, Henryk; Sigel, Roland K. O., Chem. Eur. J. 2011, 17 , 5393-5403.
With a view on protein-nucleic acid interactions in the presence of metal ions we studied the \{\textquotedbl}simple\{\textquotedbl} mixed-ligand model systems containing histamine (Ha), the metal ions Ni(2+), Cu(2+), or Zn(2+) (M(2+)), and the nucleotides adenosine 5’-triphosphate (ATP(4-)) or uridine 5’-triphosphate (UTP(4-)), which will both be referred to as nucleoside 5’-triphosphate (NTP(4-)). The stability constants of the ternary M(NTP)(Ha)(2-) complexes were determined in aqueous solution by potentiometric pH titrations. We show for both ternary-complex types, M(ATP)(Ha)(2-) and M(UTP)(Ha)(2-), that intramolecular stacking between the nucleobase and the imidazole residue occurs and that the stacking intensity is approximately the same for a given M(2+) in both types of complexes: The formation degree of the intramolecular stacks is estimated to be 20 to 50{\%}. Consequently, in protein-nucleic acid interactions imidazole-nucleobase stacks may well be of relevance. Furthermore, the well-known formation of macrochelates in binary M(2+) complexes of purine nucleotides, that is, the phosphate-coordinated M(2+) interacts with N7, is confirmed for the M(ATP)(2-) complexes. It is concluded that upon formation of the mixed-ligand complexes the M(2+)-N7 bond is broken and the energy needed for this process corresponds to the stability differences determined for the M(UTP)(Ha)(2-) and M(ATP)(Ha)(2-) complexes. It is, therefore, possible to calculate from these stability differences of the ternary complexes the formation degrees of the binary macrochelates: The closed forms amount to (65±10){\%}, (75±8){\%}, and (31±14) {\%} for Ni(ATP)(2-), Cu(ATP)(2-), and Zn(ATP)(2-), respectively, and these percentages agree excellently with previous results obtained by different methods, confirming thus the internal validity of the data and the arguments used in the evaluation processes. Based on the overall results it is suggested that M(ATP)(2-) species, when bound to an enzyme, may exist in a closed macrochelated form only, if no enzyme groups coordinate directly to the metal ion.
2010
Metal ion-N7 coordination in a ribozyme branch domain by NMR
Erat, Michèle C.; Kovacs, Helena; Sigel, Roland K. O., J. Inorg. Biochem. 2010, 104 , 611-613.
The N7 of purine nucleotides presents one of the most dominant metal ion binding sites in nucleic acids. However, the interactions between kinetically labile metal ions like Mg(2+) and these nitrogen atoms are inherently difficult to observe in large RNAs. Rather than using the insensitive direct (15)N detection, here we have used (2)J-[(1)H,(15)N]-HSQC (Heteronuclear Single Quantum Coherence) NMR experiments as a fast and efficient method to specifically observe and characterize such interactions within larger RNA constructs. Using the 27 nucleotides long branch domain of the yeast-mitochondrial group II intron ribozyme Sc.ai5gamma as an example, we show that direct N7 coordination of a Mg(2+) ion takes place in a tetraloop nucleotide. A second Mg(2+) ion, located in the major groove at the catalytic branch site, coordinates mainly in an outer-sphere fashion to the highly conserved flanking GU wobble pairs but not to N7 of the sandwiched branch adenosine.
Solution structure of a DNA double helix with consecutive metal-mediated base pairs
Johannsen, Silke; Megger, Nicole; Böhme, Dominik; Sigel, Roland K. O.; Müller, Jens, Nat. Chem. 2010, 2 , 229-234.
Metal-mediated base pairs represent a powerful tool for the site-specific functionalization of nucleic acids with metal ions. The development of applications of the metal-modified nucleic acids will depend on the availability of structural information on these double helices. We present here the NMR solution structure of a self-complementary DNA oligonucleotide with three consecutive imidazole nucleotides in its centre. In the absence of transition-metal ions, a hairpin structure is adopted with the artificial nucleotides forming the loop. In the presence of Ag(i) ions, a duplex comprising three imidazole-Ag(+)-imidazole base pairs is formed. Direct proof for the formation of metal-mediated base pairs was obtained from ¹J(¹⁵N,¹⁰⁷/¹⁰⁹Ag) couplings upon incorporation of ¹⁵N-labelled imidazole. The duplex adopts a B-type conformation with only minor deviations in the region of the artificial bases. This work represents the first structural characterization of a metal-modified nucleic acid with a continuous stretch of metal-mediated base pairs.
Controlling ribozyme activity by metal ions: Invited Review
Schnabl, Joachim; Sigel, Roland K. O., Curr. Opin. Chem. Biol. 2010, 14 , 269-275.
The observed rates of ribozyme cleavage reactions are strongly dependent on the nature of the metal ion present. Metal ions can thereby exhibit a stronger inhibiting or accelerating effect compared to Mg(2+), which is usually considered the natural cofactor. Alkaline, alkaline earth, transition, d(10), and other metal ions are applied either to gain a spectroscopic handle on the metal center, and/or to elucidate the catalytic mechanism. Here we shortly review some of the most recent publications on the influence of different metal ions on catalysis of the hammerhead, hepatitis delta virus, and group II intron ribozymes. Comparison of the observed cleavage rates of hammerhead ribozymes with the metal ion affinities of different ligands reveals that these rates correlate perfectly with the intrinsic phosphate affinities of the metal ions involved.
A stability concept for metal ion coordination to single-stranded nucleic acids and affinities of individual sites
Sigel, Roland K. O.; Sigel, Helmut, Acc. Chem. Res. 2010, 43 , 974-984.
The three-dimensional architecture and function of nucleic acids strongly depend on the presence of metal ions, among other factors. Given the negative charge of the phosphate-sugar backbone, positively charged species, mostly metal ions, are necessary for compensation. However, these ions also allow and induce folding of complicated RNA structures. Furthermore, metal ions bind to specific sites, stabilizing local motifs and positioning themselves correctly to aid (or even enable) a catalytic mechanism, as, for example, in ribozymes. Many nucleic acids thereby exhibit large differences in folding and activity depending not only on the concentration but also on the kind of metal ion involved. As a consequence, understanding the role of metal ions in nucleic acids requires knowing not only the exact positioning and coordination sphere of each specifically bound metal ion but also its intrinsic site affinity. However, the quantification of metal ion affinities toward certain sites in a single-stranded (though folded) nucleic acid is a demanding task, and few experimental data exist. In this Account, we present a new tool for estimating the binding affinity of a given metal ion, based on its ligating sites within the nucleic acid. To this end, we have summarized the available affinity constants of Mg(2+), Ca(2+), Mn(2+), Cu(2+), Zn(2+), Cd(2+), and Pb(2+) for binding to nucleobase residues, as well as to mono- and dinucleotides. We have also estimated for these ions the stability constants for coordinating the phosphodiester bridge. In this way, stability increments for each ligand site are obtained, and a clear selectivity of the ligating atoms, as well as their discrimination by different metal ions, can thus be recognized. On the basis of these data, we propose a concept that allows one to estimate the intrinsic stabilities of nucleic acid-binding pockets for these metal ions. For example, the presence of a phosphate group has a much larger influence on the overall affinity of Mg(2+), Ca(2+), or Mn(2+) compared with, for example, that of Cd(2+) or Zn(2+). In the case of Cd(2+) and Zn(2+), the guanine N7 position is the strongest intrinsic binding site. By adding up the individual increments like building blocks, one derives an estimate not only for the overall stability of a given coordination sphere but also for the most stable complex if an excess of ligating atoms is available in a binding pocket saturating the coordination sphere of the metal ion. Hence, this empirical concept of adding up known intrinsic stabilities, like building blocks, to an estimated overall stability will help in understanding the accelerating or inhibiting effects of different metal ions in ribozymes and DNAzymes.
Shaping RNA Structures with Metal Ions and Metal Ion Complexes
Sigel, Roland K. O.; Gallo, Sofia, CHIMIA 2010, 64 , 126-131.
The research in our laboratory focuses on the role of metal ions and their complexes in structure formation and folding of nucleic acids. Large catalytic RNAs, like group II introns and some riboswitches, as well as shorter RNAs and DNAs containing modified nucleotides for the assembly of nanodevices are examined. Abundant metal ions like Mg2+ or natural metabolites like coenzyme B12 are in the center of interest, but also other metal ions, complexes thereof and B12 derivatives are applied with the aim to understand the largely unknown and manifold non-covalent interactions with nucleic acids. We apply a multitude of techniques, including potentiometric pH titrations, NMR spectroscopy, X-ray crystallography, gel electrophoresis and single molecule FRET experiments. Here we briefly summarize each of our research topics emphasizing the interaction of coenzyme B12 and its derivatives with the btuB riboswitch of E. coli. This highly conserved sequence, found in the 5’-untranslated region (5’-UTR) of the btuB mRNA, is involved in the regulation of the btuB protein expression. After a summary on the historical discovery of such riboswitches and their mechanism of action, we shortly focus on our own contributions to understand the structural equilibrium, high affinity and selectivity of the interaction between this specific RNA sequence and the largest and most complex cellular metabolite, coenzyme B12.
2009
Influence of decreased solvent permittivity on the structure and magnesium(II)-binding properties of the catalytic domain 5 of a group II intron ribozyme
Furler, Maya; Knobloch, Bernd; Sigel, Roland K. O., Inorg. Chim. Acta 2009, 362 , 771-776.
Although it is well known that the so-called “equivalent solution” or “effective” solvent permittivity (dielectric constant) in proteins and nucleic acids is lower than in bulk water, this fact is commonly neglected in (bioinorganic) studies of such compounds. Using domain 5 of the group II intron ribozyme Sc.ai5γ, we describe here the influence of 1,4-dioxane-d8 on the structure and magnesium(II)-binding properties of this catalytic domain. Applying one- and two-dimensional NMR, we observe distinct structural changes in the functionally important bulge region following a decrease in solvent permittivity. Concomitantly, an increase by a factor of 1.5 in the affinity of Mg2+ towards the individual-binding sites in the catalytic core domain is observed upon addition of 1,4-dioxane-d8. This has led to the detection of a new metal ion coordination site near the GU wobble pair in the catalytic triad. Our results show that solvent permittivity is an important factor in the formation of intrinsic RNA structures and affects their metal ion-binding properties. Hence, solvent permittivity should be taken into account in future studies.
Exploring Metal Ion Coordination to Nucleic Acids by NMR
Johannsen, Silke; Korth, Maximiliane M. T.; Schnabl, Joachim; Sigel, Roland K. O., CHIMIA 2009, 63 , 146-152.
Metal ions play a crucial role in charge compensation, folding and stabilization of tertiary structures of large nucleic acids. In addition, they may be directly involved in the catalytic mechanism of ribozymes. Most metal ions applied in the context of nucleic acids in vivo and in vitro bind in a kinetically labile fashion. Hence, the detection of metal ion binding sites, not to mention the elucidation of the specific coordination sphere, still poses largely unresolved problems. Here we describe the different strategies applied and the progress made over the last years to characterize metal ion coordination to large nucleic acids by NMR.
Ca2+ induces the formation of two distinct subpopulations of group II intron molecules
Steiner, Miriam; Rueda, David; Sigel, Roland K. O., Angew. Chem. Int. Ed. 2009, 48 , 9739-9742.
New wrinkles in folding: In the folding of the D135 ribozyme derived from the groupII intron Sc.ai5γ, partial replacement of Mg2+ with Ca2+ leads to a division into two distinct subpopulations that are not interchangeable. The picture shows the splitting into the two types together with the single-molecule FRET states.
2008
Divalent metal ions tune the self-splicing reaction of the yeast mitochondrial group II intron Sc.ai5gamma
Erat, Michèle C.; Sigel, Roland K. O., J. Biol. Inorg. Chem. 2008, 13 , 1025-1036.
Group II introns are large ribozymes, consisting of six functionally distinct domains that assemble in the presence of Mg(2+) to the active structure catalyzing a variety of reactions. The first step of intron splicing is well characterized by a Michaelis-Menten-type cleavage reaction using a two-piece group II intron: the substrate RNA, the 5’-exon covalently linked to domains 1, 2, and 3, is cleaved upon addition of domain 5 acting as a catalyst. Here we investigate the effect of Ca(2+), Mn(2+), Ni(2+), Zn(2+), Cd(2+), Pb(2+), and [Co(NH(3))(6)](3+) on the first step of splicing of the Saccharomyces cerevisiae mitochondrial group II intron Sc.ai5gamma. We find that this group II intron is very sensitive to the presence of divalent metal ions other than Mg(2+). For example, the presence of only 5{\%} Ca(2+) relative to Mg(2+) results in a decrease in the maximal turnover rate k (cat) by 50{\%}. Ca(2+) thereby has a twofold effect: this metal ion interferes initially with folding, but then also competes directly with Mg(2+) in the folded state, the latter being indicative of at least one specific Ca(2+) binding pocket interfering directly with catalysis. Similar results are obtained with Mn(2+), Cd(2+), and [Co(NH(3))(6)](3+). Ni(2+) is a much more powerful inhibitor and the presence of either Zn(2+) or Pb(2+) leads to rapid degradation of the RNA. These results show a surprising sensitivity of such a large multidomain RNA on trace amounts of cations other than Mg(2+) and raises the question of biological relevance at least in the case of Ca(2+).
The corrin moiety of coenzyme B12 is the determinant for switching the btuB riboswitch of E. coli
Gallo, Sofia; Oberhuber, Michael; Sigel, Roland K. O.; Kräutler, Bernhard, ChemBioChem 2008, 9 , 1408-1414.
Riboswitches are regulatory elements in the 5’-untranslated region (5’-UTR) of bacterial mRNAs that bind certain metabolites with high specificity and affinity. The 202 nucleotide (nt)-long btuB riboswitch RNA of E. coli interacts specifically with coenzyme B12 and its derivatives thereby leading to changes in the RNA structure and hence to an altered expression of the downstream btuB gene. We report the investigations of the rearrangement of the three-dimensional structure of the btuB riboswitch upon binding to four different B12 derivatives: coenzyme B12, vitamin B12, adenosyl factor A and adenosyl-cobinamide. In-line probing experiments have shown that the corrin ring plays the crucial role in switching the three-dimensional riboswitch structure. Instead, the apical ligands influence only the binding affinity of the B12 derivative to the btuB riboswitch.
Using in vitro transcription to construct scaffolds for one-dimensional arrays of mercuric ions
Johannsen, Silke; Zelger-Paulus, Susann; Düpre, Nicole; Müller, Jens; Sigel, Roland K. O., J. Inorg. Biochem. 2008, 102 , 1141-1151.
In vitro transcription by T7 RNA polymerase can be used to construct scaffolds for the one-dimensional arrangement of mercury(II) ions. In these constructs, the metal ions are located inside of RNA double helices. By replacing the amide protons of two oppositely located uracil residues of complementary strands, mercury(II) becomes coordinated in a linear fashion to form metal-ion mediated base pairs, analogous to the well-known thymine-Hg-thymine base pair in DNA. This is shown here by a combination of various experimental techniques, including NMR spectroscopy, dynamic light scattering, as well as UV and CD spectroscopy. A total of five different double helices, including both palindromic and non-palindromic RNA sequences and between two and twenty consecutive uracil residues, have been synthesized and shown to be able to incorporate mercury(II). The synthesis of r(GGAGU 20CUCC) demonstrates that T7 polymerase is capable of handling long continuous stretches of identical nucleotides, albeit at the cost of an increasing number of abortion products and longer oligonucleotide strands that need to be separated by polyacrylamide gel electrophoresis. This work introduces RNA into the group of nucleic acids that can form metal ion mediated base pairs. The use of such metal-modified nucleic acids has been envisaged in various fields of research, including the generation of molecular wires.
Discrimination in metal-ion binding to RNA dinucleotides with a non-bridging oxygen or sulfur in the phosphate diester link
Knobloch, Bernd; Nawrot, Barbara; Okruszek, Andrzej; Sigel, Roland K. O., Chem. Eur. J. 2008, 14 , 3100-3109.
Replacement of a non-bridging oxygen in the phosphate diester bond by a sulfur has become quite popular in nucleic acid research and is often used as a probe, for example, in ribozymes, where the normally essential Mg(2+) is partly replaced by a thiophilic metal ion to reactivate the system. Despite these widely applied rescue experiments no detailed studies exist quantifying the affinity of metal ions to such terminal sulfur atoms. Therefore, we performed potentiometric pH titrations to determine the binding properties of pUp((S))U(3-) towards Mg(2+), Mn(2+), Zn(2+), Cd(2+), and Pb(2+), and compared these data with those previously obtained for the corresponding pUpU(3-) complexes. The primary binding site in both dinucleotides is the terminal phosphate group. Theoretically, also the formation of 10-membered chelates involving the terminal oxygen or sulfur atoms of the (thio)phosphate bridge is possible with both ligands. The results show that Mg(2+) and Mn(2+) exist as open (op) isomers binding to both dinucleotides only at the terminal phosphate group. Whereas Cd(pUpU)(-) only exists as Cd(pUpU)(-)(op), Cd(pUp((S))U)(-) is present to about 64 {\%} as the S-coordinated macrochelate, Cd(pUp((S))U)(-)(cl/PS). Zn(2+) forms with pUp((S))U(3-) three isomeric species, that is, Zn(pUp((S))U)(-)(op), Zn(pUp((S))U)(-)(cl/PO), and Zn(pUp((S))U)(-)(cl/PS), which occur to about 33, 12 (O-bound), and 55 {\%}, respectively. Pb(2+) forms the 10-membered chelate with both nucleotides involving only the terminal oxygen atoms of the (thio)phosphate bridge, that is, no indication of S binding was discovered in this case. Hence, Zn(2+) and Cd(2+) show pronounced thiophilic properties, whereas Mg(2+), Mn(2+), and Pb(2+) coordinate to the oxygen, macrochelate formation being of relevance with Pb(2+) only.
Divalent metal ions promote the formation of the 5’-splice site recognition complex in a self-splicing group II intron
Kruschel, Daniela; Sigel, Roland K. O., J. Inorg. Biochem. 2008, 102 , 2147-2154.
Group II introns are ribozymes occurring in genes of plants, fungi, lower eukaryotes, and bacteria. These large RNA molecular machines, ranging in length from 400 to 2500 nucleotides, are able to catalyze their own excision from pre-mRNA, as well as to reinsert themselves into RNA or sometimes even DNA. The intronic domain 1 contains two sequences (exon binding sites 1 and 2, EBS1 and EBS2) that pair with their complementary regions at the 3’-end of the 5’-exon (intron binding sites 1 and 2, IBS1 and IBS2) such defining the 5’-splice site. The correct recognition of the 5’-splice site stands at the beginning of the two steps of splicing and is thus crucial for catalysis. It is known that metal ions play an important role in folding and catalysis of ribozymes in general. Here, we characterize the specific metal ion requirements for the formation of the 5’-splice site recognition complex from the mitochondrial yeast group II intron Sc.ai5gamma. Circular dichroism studies reveal that the formation of the EBS1.IBS1 duplex does not necessarily require divalent metal ions, as large amounts of monovalent metal ions also promote the duplex, albeit at a 5000 times higher concentration. Nevertheless, micromolar amounts of divalent metal ions, e.g. Mg2+ or Cd2+, strongly promote the formation of the 5’-splice site. These observations illustrate that a high charge density independent of the nature of the ion is needed for binding EBS1 to IBS1, but divalent metal ions are presumably the better players.
Effect of the ribose versus 2’-deoxyribose residue on the metal ion-binding properties of purine nucleotides
Mucha, Ariel; Knobloch, Bernd; Jeżowska-Bojczuk, Małgorzata; Kozłowski, Henryk; Sigel, Roland K. O., Dalton Trans. 2008, 0 , 5368-5377.
The interaction between metal ions and nucleotides is well characterized, as is their importance for metabolic processes, e.g. in the synthesis of nucleic acids. Hence, it is surprising to find that no detailed comparison is available of the metal ion-binding properties between nucleoside 5’-phosphates and 2’-deoxynucleoside 5’-phosphates. Therefore, we have measured here by potentiometric pH titrations the stabilities of several metal ion complexes formed with 2’-deoxyadenosine 5’-monophosphate (dAMP2-), 2’-deoxyadenosine 5’-diphosphate (dADP3-) and 2’-deoxyadenosine 5’-triphosphate (dATP4-). These results are compared with previous data measured under the same conditions and available in the literature for the adenosine 5’-phosphates, AMP(2-), ADP(3-) and ATP(4-), as well as guanosine 5’-monophosphate (GMP(2-)) and 2’-deoxyguanosine 5’-monophosphate (dGMP(2-)). Hence, in total four nucleotide pairs, GMP(2-)/dGMP(2-), AMP(2-)/dAMP(2-), ADP(3-)/dADP(3-) and ATP(4-)/dATP(4-) (= NP/dNP), could be compared for the four metal ions Mg2+, Ni2+, Cu2+ and Zn2+ (= M2+). The comparisons show that complex stability and extent of macrochelate formation between the phosphate-coordinated metal ion and N7 of the purine residue is very similar (or even identical) for the AMP(2-)/dAMP(2-) and ADP(3-)/dADP(3-) pairs. In the case of the complexes formed with ATP(4-)/dATP(4-) the 2’-deoxy complexes are somewhat more stable and show also a slightly enhanced tendency for macrochelate formation. This is different for guanine nucleotides: the stabilities of the M(dGMP) complexes are clearly higher, as are the formation degrees of their macrochelates, than is the case with the M(GMP) complexes. This enhanced complex stability and greater tendency to form macrochelates can be attributed to the enhanced basicity (DeltapKaca. 0.2) of N7 in the 2’-deoxy compound. These results allow general conclusions regarding nucleic acids to be made.
Comparison of the acid-base properties of ribose and 2’-deoxyribose nucleotides
Mucha, Ariel; Knobloch, Bernd; Jeżowska-Bojczuk, Małgorzata; Kozłowski, Henryk; Sigel, Roland K. O., Chem. Eur. J. 2008, 14 , 6663-6671.
The extent to which the replacement of a ribose unit by a 2’-deoxyribose unit influences the acid-base properties of nucleotides has not hitherto been determined in detail. In this study, by potentiometric pH titrations in aqueous solution, we have measured the acidity constants of the 5’-di- and 5’-triphosphates of 2’-deoxyguanosine [i.e., of H(2)(dGDP)(-) and H(2)(dGTP)(2-)] as well as of the 5’-mono-, 5’-di-, and 5’-triphosphates of 2’-deoxyadenosine [i.e., of H(2)(dAMP)(+/-), H(2)(dADP)(-), and H(2)(dATP)(2-)]. These 12 acidity constants (of the 56 that are listed) are compared with those of the corresponding ribose derivatives (published data) measured under the same experimental conditions. The results show that all protonation sites in the 2’-deoxynucleotides are more basic than those in their ribose counterparts. The influence of the 2’-OH group is dependent on the number of 5’-phosphate groups as well as on the nature of the purine nucleobase. The basicity of N7 in guanine nucleotides is most significantly enhanced (by about 0.2 pK units), while the effect on the phosphate groups and the N1H or N1H(+) sites is less pronounced but clearly present. In addition, (1)H NMR chemical shift change studies in dependence on pD in D(2)O have been carried out for the dAMP, dADP, and dATP systems, which confirmed the results from the potentiometric pH titrations and showed the nucleotides to be in their anti conformations. Overall, our results are not only of relevance for metal ion binding to nucleotides or nucleic acids, but also constitute an exact basis for the calculation, determination, and understanding of perturbed pK(a) values in DNAzymes and ribozymes, as needed for the delineation of acid-base mechanisms in catalysis.
2007
Solution structure of domain 6 from a self-splicing group II intron ribozyme: A Mg(2+) binding site is located close to the stacked branch adenosine
Erat, Michèle C.; Zerbe, Oliver; Fox, Thomas; Sigel, Roland K. O., ChemBioChem 2007, 8 , 306-314.
Group II intron self-splicing is essential for the correct expression of organellar genes in plants, fungi, and yeast, as well as of bacterial genes. Self-excision of these autocatalytic introns from the primary RNA transcript is achieved in a two-step mechanism that is apparently analogous to that of the eukaryotic spliceosome. The 2’-OH of a conserved adenosine (the branch point) located within domain 6 (D6) acts as the nucleophile in the first step of splicing. Despite the biological importance of group II introns, little is known about their structural organization and usage of metal ions in catalysis. Here we report the first solution structure of a catalytically active D6 construct encompassing the branch point and the neighboring helical regions from the mitochondrial yeast intron ai5gamma. The branch adenosine is the single unpaired nucleotide, and, in contrast to the spliceosomal branch site, resides within the helix, being partially stacked between two flanking GU wobble pairs. We identified a novel prominent Mg(2+) binding site in the major groove of the branch site. Importantly, Mg(2+) addition does not impair the stacking of the branch adenosine, rather it strengthens the interaction with the flanking uridines, as shown by NMR and fluorescence studies. This means that domain 6 presents the branch adenosine in a stacked fashion to the core of group II introns upon folding to the active conformation.
Determination of the intrinsic affinities of multiple site-specific Mg(2+) ions coordinated to domain 6 of a group II intron ribozyme
Erat, Michèle C.; Sigel, Roland K. O., Inorg. Chem. 2007, 46 , 11224-11234.
Group II introns are large metallo-ribozymes that use divalent metal ions in folding and catalysis. The 3’-terminal domain 6 (D6) contains a conserved adenosine whose 2’-OH group acts as the nucleophile in the first splicing step. In the hierarchy of folding, D6 binds last into the active site. In order to investigate and understand the folding process to the catalytically active intron structure, it is important to know the individual binding affinities of Mg2+ ions to D6. We recently studied the solution structure of a 27 nucleotide long D6 (D6-27) from the mitochondrial yeast group II intron Sc.ai5gamma, also identifying five Mg2+ binding sites including the one at the 5’-terminal phosphate residues. Mg2+ coordination to the 5’-terminal di- and triphosphate groups is strongest (e.g., log KA,TP = 4.55 +/- 0.10) and is evaluated here in detail for the first time. The other four binding sites within D6-27 are filled simultaneously (e.g., log KA,BR = 2.38 +/- 0.06) and thus compete for the free Mg2+ ions in solution, having a distinct influence on the individual affinities of the various sites. For the first time, we take this competition into account to obtain the intrinsic binding constants, describing a method that is generally applicable. Our data illustrates that any RNA molecule undergoing tertiary contacts to a second RNA molecule first needs to be loaded evenly and specifically with metal ions to compensate for the repulsion between the negatively charged RNA molecules.
From nucleotides to ribozymes—A comparison of their metal ion binding properties: Review
Freisinger, Eva; Sigel, Roland K. O., Coord. Chem. Rev. 2007, 251 , 1834-1851.
It is undisputable that the fates of metal ions and nucleic acids are inescapably interwoven. Metal ions are essential for charge compensation of the negatively charged phosphate–sugar backbone, they are instrumental for proper folding, and last but not least they are crucial cofactors for ribozyme catalysis. Considerable progress has been achieved in the past few years on the identification of metal ion binding sites in large DNA and RNA molecules, like in ribozymes including the ribosome. Hereby, most information was gained from crystallography, which fails to explain metal ion binding equilibria in solution as well as the factors that determine the coordination of a metal ion to a specific site. In contrast, such information is readily available for the low-molecular building blocks of large nucleic acids, i.e. for mononucleotides and to some extent also dinucleotides. In this review, we combine and compare for the first time both sets of information. The focus is thereby set on Mg2+, Ca2+, Mn2+, and Cd2+ because these four metal ions are either freely available in cells, have a large impact on the catalytic rate of ribozymes, and/or are often applied in RNA biochemistry. Our comparisons show that results obtained from small molecules can be directly transposed to the findings in large RNA structures like the ribosome. For example, the basic coordination-chemical properties of the different metal ions are reflected in their binding to large nucleic acid structures: macrochelate formation, e.g. the simultaneous intranucleotide coordination of a Mg2+ ion to the phosphate unit and the N7 site of a purine nucleobase (be it inner- or outersphere), is well known for mononucleotides. We show that the frequency of occurrence of this type of coordination is the same for mononucleotides and the ribosome.
Towards the synthesis of light-stable coenzyme B12 analogs
Gallo, Sofia; Freisinger, Eva; Sigel, Roland K. O., Inorg. Chim. Acta 2007, 360 , 360-368.
The organometallic complex coenzyme B12 (adenosyl cobalamin, AdoCbl) is not only an essential coenzyme in many biochemical reactions of most if not all living organisms but has lately been shown to play a crucial role in the regulation of B12 related genes. As a consequence, coenzyme B12 has been a target of intense research. However, the investigations of AdoCbl have often been hampered due to its high light-sensitivity leading to decomposition of the compound within a few seconds. Here, we describe a strategy to synthesize more light-stable coenzyme B12 analogs, which show similar steric properties as adenosyl cobalamin. The synthesis, structural characterization as well as the pH dependent “base-on/base-off” behavior of cyanide bridged vitamin B12 conjugates with either a cis-[(NH3)2Pt]2+ or an [enPt]2+ moiety, leading to cis-[(NH3)2PtCl-vitB12]+ (1) and [enPtCl-vitB12]+ (2) are reported. The subsequent reaction of cis-[(NH3)2PtCl-vitB12]+ with the model nucleobase 9-methyladenine leads to the corresponding adduct, where the adenine moiety is coordinated to the Pt2+ center either via N1 or N7. This compound is light-stable and harbors the adenine moiety in the same distance of 5 Å above the corrin plane as present in the highly light-sensitive adenosyl cobalamin.
Metal-ion-coordinating properties of the dinucleotide 2’-deoxyguanylyl(5’‐3’)-2’-deoxy-5’-guanylate (d(pGpG)3-): Isomeric equilibria including macrochelated complexes relevant for nucleic acids
Knobloch, Bernd; Sigel, Helmut; Okruszek, Andrzej; Sigel, Roland K. O., Chem. Eur. J. 2007, 13 , 1804-1814.
The interaction between divalent metal ions and nucleic acids is well known, yet knowledge about the strength of binding of labile metal ions at the various sites is very scarce. We have therefore studied the stabilities of complexes formed between the nucleic acid model d(pGpG) and the essential metal ions Mg2+ and Zn2+ as well as with the generally toxic ions Cd2+ and Pb2+ by potentiometric pH titrations; all four ions are of relevance in ribozyme chemistry. A comparison of the present results with earlier data obtained for M(pUpU)- complexes allows the conclusion that phosphate-bound Mg2+ and Cd2+ form macrochelates by interaction with N7, whereas the also phosphate-coordinated Pb2+ forms a 10-membered chelate with the neighboring phosphate diester bridge. Zn2+ forms both types of chelates with formation degrees of about 91{\%} and 2.4{\%} for Zn[d(pGpG)]cl/N7 and Zn[d(pGpG)]-cl/PO, respectively; the open form with Zn2+ bound only to the terminal phosphate group, Zn[d(pGpG)]-op, amounts to about 6.8 {\%}. The various intramolecular equilibria have also been quantified for the other metal ions. Zn2+, Cu2+, and Cd2+ also form macrochelates in the monoprotonated M[H;d(pGpG)] species (the proton being at the terminal phosphate group), that is, the metal ion at N7 interacts to some extent with the P(O)2(OH)- group. Thus, this study demonstrates that the coordinating properties of the various metal ions toward a pGpG unit in a nucleic acid differ: Mg2+, Zn2+, and Cd2+ have a significant tendency to bridge the distance between N7 and the phosphate group of a (d)GMP unit, although to various extents, whereas Pb2+ (and possibly Ca2+) prefer a pure phosphate coordination.
Alternative roles for metal ions in enzyme catalysis and the implications for ribozyme chemistry - Review
Sigel, Roland K. O.; Pyle, Anna Marie, Chem. Rev. 2007, 107 , 97-113.
This is a Review with no abstract
Binding interaction of Re(H2O)3(CO)3+ with the DNA fragment d(CpGpG)
Zobi, Fabio; Blacque, Olivier; Sigel, Roland K. O.; Alberto, Roger, Inorg. Chem. 2007, 46 , 10458-10460.
Insights into the interaction of the [Re(H2O)3(CO)3]+ complex (1) with the DNA fragment d(CpGpG) have been obtained by one- (1D) and two-dimensional (2D) NMR spectroscopy. The H8 resonances of the single major [Re(H2O)d(CpGpG)(CO)3]- adduct (2) exhibit pH-independent chemical shift changes attributable to metal N7 binding. The structure of this adduct has been characterized by molecular modeling studies based on 1D and 2D NMR data. In solution, 2 shows the presence of two N7-coordinated guanine moieties in a head-to-head (HH) orientation as evidenced by G2H8/G3H8 cross-peaks in the 1H-1H NOESY NMR spectrum. The presence of the 5’-bridging phosphodiester appears to stabilize the HH1 L conformer, as was previously described for related Pt and Rh complexes.
2006
Acid-base properties of the nucleic-acid model 2’-deoxyguanylyl(5’‐3’)-2’-deoxy-5’-guanylate, d(pGpG)3-, and of related guanine derivatives
Knobloch, Bernd; Sigel, Helmut; Okruszek, Andrzej; Sigel, Roland K. O., Org. Biomol. Chem. 2006, 4 , 1085-1090.
The dinucleotide d(pGpG) is an often employed DNA model to study various kinds of interactions between DNA and metal ions, but its acid-base properties were not yet described in detail. In this study the six deprotonation reactions of H4[d(pGpG)]+ are quantified. The acidity constants for the release of the first proton from the terminal P(O)(OH)2 group (pKa = 0.65) and for one of the (N7)H+ sites (pKa = 2.4) are estimated. The acidity constants of the remaining four deprotonation reactions were measured by potentiometric pH titrations in aqueous solution (25 degrees C; I = 0.1 M, NaNO3): The pKa values for the deprotonations of the second (N7)H+, the P(O)2(OH)-, and the two (N1)H sites are 2.98, 6.56, 9.54 and 10.11, respectively. Based on these results we show how to estimate acidity constants for related systems that have not been studied, e.g. pGpG, which is involved in the initiation step of a rotavirus RNA polymerase. The relevance of our results for nucleic acids in general is briefly indicated.
Pyrazine as a building block for molecular architectures with PtII
Willermann, Michael; Mulcahy, Clodagh; Sigel, Roland K. O.; Cerdà, Marta Morell; Freisinger, Eva; Sanz Miguel, Pablo J.; Roitzsch, Michael; Lippert, Bernhard, Inorg. Chem. 2006, 45 , 2093-2099.
A series of pyrazine (pz) complexes containing cis-(NH(3))(2)Pt(II), (tmeda)Pt(II) (tmeda = N,N,N’,N’-tetramethylethylenediamine), and trans-(NH(3))(2)Pt(II) entities have been prepared and characterized by X-ray crystallography and/or 1H NMR spectroscopy. In these compounds, the pz ligands act as monodentate (1-3) or bidentate bridging ligands (4-7). Three variants of the latter case are described: a dinuclear complex [Pt(II)]2 (4b), a cyclic tetranuclear [Pt(II)](4) complex (5), and a trinuclear mixed-metal complex [Pt2Ag] (7). Mono- and bidentate binding modes are readily differentiated by 1H NMR spectroscopy, and the assignment of pz protons in the case of monodentate coordination is aided by the observation of (195)Pt satellites. Formation of the open molecular box cis-[(NH3)2Pt(pz)4](NO3)8.3.67H2O (5) from cis-(NH3)2Pt(II) and pz follows expectations of the \{\textquotedbl}molecular library approach\{\textquotedbl} for the generation of a cyclic tetramer.
2005
Structure Determination of Catalytic RNAs and Investigations of Their Metal Ion-Binding Properties
Erat, Michèle C.; Sigel, Roland K. O., CHIMIA 2005, 59 , 817-821.
Naturally occurring RNA molecules exhibit many unexpected and fascinating properties in living cells such as protein synthesis and transport, regulation of metabolic functions, and catalytic cleavage reactions. To understand this functional diversity, a detailed knowledge of RNA structure and metal ion-binding properties is crucial. In our research group, we address these problems by combining various biochemical, analytical and spectroscopic techniques. A large part of our work is devoted to the structure determination of catalytic RNA molecules, i.e. ribozymes, by NMR. Based on the three-dimensional structure, further experiments are carried out to understand in detail the effects of different metal ions on the local and global structure, as well as catalysis itself.
In vitro Transcription and Purification of RNAs of Different Size
Gallo, Sofia; Furler, Maya; Sigel, Roland K. O., CHIMIA 2005, 59 , 812-816.
In vitro run-off transcription from a double-stranded DNA template by T7 RNA polymerase is an elegant way to obtain highly pure and uniform RNA oligonucleotides of lengths ranging from about 15 to several thousand nucleotides. Here we describe the different strategies applied and optimized in our laboratory to enzymatically synthesize RNAs as necessary when working at the interface of bioinorganic chemistry, coordination chemistry, RNA biochemistry and structural biology.
Acid-base and metal-ion binding properties of the RNA dinucleotide uridylyl-(5’‐3’)-5’uridylate (pUpU3-)
Knobloch, Bernd; Suliga, Danuta; Okruszek, Andrzej; Sigel, Roland K. O., Chem. Eur. J. 2005, 11 , 4163-4170.
It is well known that Mg2+ and other divalent metal ions bind to the phosphate groups of nucleic acids. Subtle differences in the coordination properties of these metal ions to RNA, especially to ribozymes, determine whether they either promote or inhibit catalytic activity. The ability of metal ions to coordinate simultaneously with two neighboring phosphate groups is important for ribozyme structure and activity. However, such an interaction has not yet been quantified. Here, we have performed potentiometric pH titrations to determine the acidity constants of the protonated dinucleotide H2(pUpU)-, as well as the binding properties of pUpU3- towards Mg2+, Mn2+, Cd2+, Zn2+, and Pb2+. Whereas Mg2+, Mn2+, and Cd2+ only bind to the more basic 5’-terminal phosphate group, Pb2+, and to a certain extent also Zn2+, show a remarkably enhanced stability of the [M(pUpU)]- complex. This can be attributed to the formation of a macrochelate by bridging the two phosphate groups within this dinucleotide by these metal ions. Such a macrochelate is also possible in an oligonucleotide, because the basic structural units are the same, despite the difference in charge. The formation degrees of the macrochelated species of [Zn(pUpU)]- and [Pb(pUpU)]- amount to around 25 and 90 {\%}, respectively. These findings are important in the context of ribozyme and DNAzyme catalysis, and explain, for example, why the leadzyme could be selected in the first place, and why this artificial ribozyme is inhibited by other divalent metal ions, such as Mg2+.
Group II Intron Ribozymes and Metal Ions - A Delicate Relationship: Microreview
Sigel, Roland K. O., Eur. J. Inorg. Chem. 2005, 2005 , 2281-2292.
Group II introns are naturally occurring ribozymes in plants, fungi, bacteria, and lower eukaryotes that undergo a fascinating array of reactions. These large molecular machines with a size ranging between 600 and 2500 nucleotides are self-splicing introns also capable of reinserting themselves into RNA or DNA, thus making them mobile genetic elements. The structural information available on group II intron ribozymes is very scarce. So far, only one crystal structure and one NMR solution structure of two domains located in the catalytic core are available. For proper folding and function, each intron requires specific concentrations of monovalent and divalent metal ions. Although most of these metal ions are used for charge screening, some are bound to distinct sites as has been shown by hydrolytic cleavage experiments. These specifically bound ions are crucial for tertiary contact formation and catalysis. This review will discuss the different metal-ion requirements of self-splicing group II introns, the available structural data and information on the binding location and affinity of metal ions, as well as the methods applied to investigate the metal-ion binding properties of these large RNAs. Due to the size of these introns, the richness of local structures, the catalytic versatility and the involvement of metal ions in all of the above-mentioned aspects, group II introns are an ideal target to be studied by combined means from the fields of Biochemistry, Molecular Biology, Analytical, and (Bio)Inorganic Chemistry. (© Wiley-VCH Verlag GmbH {\&} Co. KGaA, 69451 Weinheim, Germany, 2005)
2004
Two metal ions coordinated to a purine residue tolerate each other well
Knobloch, Bernd; Sigel, Roland K. O.; Lippert, Bernhard; Sigel, Helmut, Angew. Chem. Int. Ed. 2004, 43 , 3793-3795.
The catalytic sites of ribozymes generally contain two or more metal ions. Guanine is the nucleobase most often involved in binding to metal ions. The repulsion between two M2+ ions simultaneously coordinated to guanine in aqueous solution was investigated by using complex 1 (see picture; dien=diethylenetriamine, R=C2H5) as a model, for which the stability constants for the binding of Mg2+ and Cu2+ were determined.
Solution structure of domain 5 of a group II intron ribozyme reveals a new RNA motif
Sigel, Roland K. O.; Sashital, Dipali G.; Abramovitz, Dana L.; Palmer, Arthur G.; Butcher, Samuel E.; Pyle, Anna Marie, Nat. Struct. Mol. Biol. 2004, 11 , 187-192.
Domain 5 (D5) is the central core of group II intron ribozymes. Many base and backbone substituents of this highly conserved hairpin participate in catalysis and are crucial for binding to other intron domains. We report the solution structures of the 34-nucleotide D5 hairpin from the group II intron ai5 gamma in the absence and presence of divalent metal ions. The bulge region of D5 adopts a novel fold, where G26 adopts a syn conformation and flips down into the major groove of helix 1, close to the major groove face of the catalytic AGC triad. The backbone near G26 is kinked, exposing the base plane of the adjacent A-U pair to the solvent and causing bases of the bulge to stack intercalatively. Metal ion titrations reveal strong Mg(2+) binding to a minor groove shelf in the D5 bulge. Another distinct metal ion-binding site is observed along the minor groove side of the catalytic triad, in a manner consistent with metal ion binding in the ribozyme active site.
2002
N1 and N3 Linkage Isomers of Neutral and Deprotonated Cytosine with trans-[(CH3NH2)2PtII]
Brüning, Wolfgang; Freisinger, Eva; Sabat, Michal; Sigel, Roland K. O.; Lippert, Bernhard, Chem. Eur. J. 2002, 8 , 4681-4692.
A series of complexes obtained from the reaction of trans-[(CH3NH2)2PtII] with unsubstituted cytosine (CH) and its anion (C), respectively, has been prepared and isolated or detected in solution: trans-[Pt(CH3NH2)2(CH-N3)Cl]Cl⋅H2O (1), trans-[Pt(CH3NH2)2(CH-N3)2](ClO4)2 (1 a), trans-[Pt(CH3NH2)2(C-N3)2]⋅2 H2O (1 b), trans-[Pt(CH3NH2)2(CH-N3)2](ClO4)2⋅2 DMSO (1 c), trans-[Pt(CH3NH2)2(CH-N1)2](NO3)2⋅3 H2O (2 a), trans-[Pt(CH3NH2)2(C-N1)2]⋅2 H2O (2 b), trans-[Pt(CH3NH2)2(CH-N1)(CH-N3)](ClO4)2 (3 a), trans-[Pt(CH3NH2)2(C-N1)(C-N3)] (3 b), and trans-[Pt(CH3NH2)2(N1-C-N3)(N3-C-N1)Cu(OH)]ClO4⋅1.2 H2O (4). X-ray crystal structures of all these compounds, except 3 a and 3 b, are reported. Complex 2 a is of particular interest in that it contains the rarer of the two 2-oxo-4-amino tautomer forms of cytosine, namely that with the N3 position protonated. Since the effect of PtII on the geometry of the nucleobase is minimal, bond lengths and angles of CH in 2 a reflect, to a first approximation, those of the free rare tautomer. Compared to the preferred 2-oxo-4-amino tautomer (N1 site protonated) of CH, the rare tautomer in 2 a differs particularly in internal ring angles (7–11 σ). Formation of compounds containing the rare CH tautomers on a preparative scale can be achieved by a detour (reaction of PtII with the cytosine anion, followed by cytosine reprotonation) or by linkage isomerization (N3→N1) under alkaline reaction conditions. Surprisingly, in water and over a wide pH range, N1 linkage isomers (3 a, 2 a) form in considerably higher amounts than can be expected on the basis of the tautomer equilibrium. This is particularly true for the pH range in which the cytosine is present as a neutral species and implies that complexation of the minor tautomer is considerably promoted. Deprotonation of the rare CH tautomers in 2 a occurs with pKa values of 6.07±0.18 (1 σ) and 7.09±0.11 (1 σ). This value compares with pKa 9.06±0.09 (1 σ) (average of both ligands) in 1 a.
Hydrogen bonding patterns of 7,9-dimethylguanine and its transplatinum(II) complexes
Sigel, Roland K. O.; Freisinger, Eva; Abbate, Michele; Lippert, Bernhard, Inorg. Chim. Acta 2002, 339 , 355-365.
Methylation at the N7 position is one of the most frequently naturally occurring modifications of guanosine. This alteration drastically changes the hydrogen bonding and acid–base properties of the guanine nucleobase. Here we show on the example of the model nucleobase 7,9-dimethylguanine that due to blockage of N7 of the purine ring, new hydrogen bonding patterns occur on the minor groove binding face of this nucleobase involving the ring nitrogen N3 and the exocyclic amino group N2H2. The free 7,9-dimethylguaninium ion and several transplatinum(II) complexes of the this ligand are presented and discussed.
2001
PtII Binding to N1 of Cytosine: Strengthening the Watson-Crick Pair with Guanine and yet Confining Its pH Existence Range
Brüning, Wolfgang; Sigel, Roland K. O.; Freisinger, Eva; Lippert, Bernhard, Angew. Chem. Int. Ed. 2001, 40 , 3397-3399.
Metal coordination to a nucleobase donor atom remote from its hydrogen bonding sites neither prevents base-pair formation for steric reasons nor diminishes their strength. On the contrary, it enhances it. This effect, which was originally proposed by theoretical chemists and attributed to polarization effects of the metal ion as well as electrostatic attraction,1 has recently been found to be also true for solutions of guanine nucleobases carrying a PtII entity at N7.2 X-ray crystallographic results are consistent with this view (2a, 3). As will be shown here, the previously demonstrated case of Watson–Crick pairing between N7-platinated guanine and cytosine can be inverted by coordination of the metal to the cytosine N1 position and hydrogen bonding it with a nonmetalated guanine (Scheme 1). The order of stability in DMSO solution is II≈III>I; thus, the artificial (platinated) base pairs are more stable than the natural ones.
Metal-Modified Nucleobase Sextet: Joining Four Linear Metal Fragments (trans-a2PtII) and Six Model Nucleobases to an Exceedingly Stable Entity
Sigel, Roland K. O.; Thompson, Susan M.; Freisinger, Eva; Glahé, Frank; Lippert, Bernhard, Chem. Eur. J. 2001, 7 , 1968-1980.
Crosslinking of three different model nucleobases (9-ethyladenine, 9-EtA; 9-ethylguanine, 9-EtGH; 1-methyluracil, 1-MeU) by two linear trans-a2PtII (a= NH3 or CH3NH2) entities leads to a flat metal-modified base triplet, trans,trans-[(NH3)2Pt(1-MeU-N3)(μ-9-EtA-N7,N1)Pt(CH3NH2)2(9-EtGH-N7)]3+ (4 b). Upon hemideprotonation of the 9-ethylguanine base at the N1 position, 4 b spontaneously dimerizes to the metalated nucleobase sextet 5, [(4 b)≡(4 b-H)]5+. In this dimeric structure a neutral and an anionic guanine ligand, which are complementary to each other, are joined through three H bonds and additionally by two H bonds between guanine and uracil nucleobases. Four additional interbase H bonds maintain the approximate coplanarity of all six bases. The two base triplets form an exceedingly stable entity (KD=500±150 M−1 in DMSO), which is unprecedented in nucleobase chemistry. The precursor of 4 b and several related complexes are described and their structures and solution properties are reported.
2000
Heavy metal mutagenicity: insights from bioinorganic model chemistry
Müller, Jens; Sigel, Roland K. O.; Lippert, Bernhard, J. Inorg. Biochem. 2000, 79 , 261-265.
The mutagenicity of metal species may be the result of a direct interaction with the target molecule DNA. Possible scenarios leading to nucleobase mispairing are discussed, and selected examples are presented. They include changes in nucleobase selectivity as a consequence of alterations in acid–base properties of nucleobase atoms and groups involved in complementary H bond formation, guanine deprotonation, and stabilization of rare nucleobase tautomers by metal ions. Oxidative nucleobase damage brought about by metal species will not be considered.
Hydrogen bonding between adenine and 2,4-difluorotoluene is definitely not present, as shown by concentration-dependent NMR studies
Schmidt, Kathrin S.; Sigel, Roland K. O.; Filippov, Dmitri V.; van der Marel, Gijs A.; Lippert, Bernhard; Reedijk, Jan, New J. Chem. 2000, 24 , 195-197.
The chloroform-soluble nucleobase derivatives N9-cyclohexylmethyladenine (A) and N1-cyclohexylmethylthymine (T) have been synthesized in order to study hydrogen-bonding interactions between A and the thymidine mimic 2,4-difluorotoluene (F) in CDCl3 at high concentrations. Concentration-dependent 1H NMR experiments show that in the presence of F, A undergoes self-association rather than pairing with F. These results strongly support the assumptions made by Kool with regard to the lack of hydrogen bonding between adenine and 2,4-difluorotoluene.
Metal ion binding sites in a group II intron core
Sigel, Roland K. O.; Vaidya, Anand; Pyle, Anna Marie, Nat. Struct. Biol. 2000, 7 , 1111-1116.
Group II introns are catalytic RNA molecules that require divalent metal ions for folding, substrate binding, and chemical catalysis. Metal ion binding sites in the group II core have now been elucidated by monitoring the site-specific RNA hydrolysis patterns of bound ions such as Tb(3+) and Mg(2+). Major sites are localized near active site elements such as domain 5 and its surrounding tertiary interaction partners. Numerous sites are also observed at intron substructures that are involved in binding and potentially activating the splice sites. These results highlight the locations of specific metal ions that are likely to play a role in ribozyme catalysis.
Effects of N7-methylation, N7-platination, and C8-hydroxylation of guanine on H-bond formation with cytosine: Platinum coordination strengthens the Watson-Crick pair
Sigel, Roland K. O.; Freisinger, Eva; Lippert, Bernhard, J. Biol. Inorg. Chem. 2000, 5 , 287-299.
The hydrogen bonding properties of 1-methylcytosine (1-MeC) with the following guanine base derivatives have been studied in DMSO-d6, applying concentration-dependent 1H NMR spectroscopy: 9-ethylguanine, 7,9-dimethylguanine (7,9-DimeGH+), and 7,8-dihydro-8-oxo-9-methylguanine (8-O-9-MeGH), as well as three 9-ethylguanine complexes carrying different Pt(II) moieties at the N7 position. The association constants K for the Watson-Crick pairing schemes are by a factor 2–3 higher in the cases of platinated guanine complexes compared to the Watson-Crick pair between 9-ethylguanine and 1-methylcytosine (K= 6.9 ± 1.3 M−1). Similar enhanced stabilities are observed for the pairs formed between 1-MeC and 7,9-DimeGH+ or 8-O-9-MeGH. The increase in N1H acidity of the guanine derivative upon modification at the N7 or C8 positions can be correlated with the association constants K; the result is a bell-shaped curve meaning that acidification initially stabilizes hydrogen bond formation up to a certain maximum; further acidification then leads to a destabilization. For two of the examples studied in solution, hydrogen bonding according to Watson-Crick between N7-platinated 9-ethylguanine and 1-methylcytosine has also been established by X-ray crystallography.
1999
Combining four different model nucleobases (uracil, adenine, guanine, cytosine) via metal binding and H bond formation in a single compound
Sigel, Roland K. O.; Thompson, Susan M.; Freisinger, Eva; Lippert, Bernhard, Chem. Commun. 1999, 0 , 19-20.
A cyclic arrangement of the four model nucleobases 1-methyluracilate (mura), 9-ethyladenine (eade), 9-ethylguanine (Hegua) and 1-methylcytosine (mcyt), held together by two metal entities [trans-(NH3)2PtII and trans-(MeNH2)2PtII] and multiple H bond interactions, is presented.
Metal-Modified Base Pairs Involving Different Donor Sites of Purine Nucleobases: Trans -[a 2 Pt(7,9-DimeG- N1 )(9-EtGH- N7 )] 2+ and trans -[a 2 Pt(7,9-DimeG- N1 )(9-EtG- N7 )] + (a = NH 3 or CH 3 NH 2 ; 9-EtGH = 9-Ethylguanine; 7,9-DimeG = 7,9-Dimethylguanine). Possible Relevance to Metalated DNA Triplex Structures
Sigel, Roland K. O.; Sabat, Michal; Freisinger, Eva; Mower, Amanda; Lippert, Bernhard, Inorg. Chem. 1999, 38 , 1481-1490.
The X-ray crystal structures of four mixed-nucleobase complexes 1a, 1b, 2, and 4 of trans-a2PtII (a = NH3 or CH3NH2) with N1-bonded 7,9-dimethylguanine (7,9-DimeG) and N7-bonded 9-ethylguanine (9-EtGH) are reported. The compounds are discussed in terms of their possible model character for platinated nucleobase triplets of the pu × pu·pym (pu = purine, pym = pyrimidine) type and are compared with known guanine·guanine pairs. Structural differences resulting from a “metal modification” have been studied, as have been stacking and H bonding motifs. 1a and 1b are two modifications of the same compound, trans-[(NH3)2Pt(7,9-DimeG)(9-EtGH)](ClO4)2·H2O. X-ray data for 1a: orthorhombic, Pca21, a = 13.833(3) Å, b = 14.357(3) Å, c = 13.293(3) Å, Z = 4, R = 0.027. 1b: monoclinic, P21/n, a = 13.987(3) Å, b = 12.582(3) Å, c = 15.613(3) Å, β = 104.61(3)°, Z = 4, R = 0.034. trans-[(CH3NH2)2Pt(7,9-DimeG)(9-EtGH)](ClO4)2 (2): orthorhombic, Pca21, a = 13.886(1) Å, b = 14.698(1) Å, c = 13.008(3) Å, Z = 4, R = 0.034. trans-[a2Pt(7,9-DimeG)(9-EtG)](ClO4)·nH2O (3, a = NH3, n = 2; 4, a = CH3NH2, n = 3). X-ray data for 4: monoclinic, C2/c, a = 23.296(5) Å, b = 10.566(2) Å, c = 22.327(4) Å, β = 93.73(3)°, Z = 8, R = 0.047. The metalated base triplet 2·(1-MeC) (1-MeC = 1-methylcytosine) has been detected in DMSO-d6 solution through 1H NMR spectroscopy, and an ab initio optimized structure of an analogous platinated base triplet has been obtained.
PtII coordination to guanine-N7: enhancement of the stability of the Watson-Crick base pair with cytosine
Sigel, Roland K. O.; Lippert, Bernhard, Chem. Commun. 1999, 0 , 2167-2168.
Concentration-dependent 1H NMR shift experiments in (CD3)2SO reveal that PtII-coordination to N7 of guanine enhances the stability of the Watson–Crick pair with cytosine.
1998
Creating regular arrangements of nucleobases through metal ion coordination and H bond formation
Lippert, Bernhard; Amo-Ochoa, Pilar; Brüning, Wolfgang; Freisinger, Eva; Lüth, Marc-Sven; Meier, Susanne; Meiser, Cordula; Metzger, Susanne; Rauter, Holger; Schreiber, Andre; Sigel, Roland K. O.; Witkowski, Holger, Pure Appl. Chem. 1998, 70 , 977-983.
If interactions between nucleobases are extened beyond the normal H bonding schemes by also allowing covalent metal ion cross-linking, complexes of varying topologies are created, which open the field of metal-nucleic acid chemistry from biologically relevant model compound to molecular recognition and supramolecular chemistry.
Metalated Nucleobase Quartets: Dimerization of a Metal-Modified Guanine, Cytosine Pair of trans-(NH3)2PtII and Formation of CH···N Hydrogen Bonds
Sigel, Roland K. O.; Freisinger, Eva; Metzger, Susanne; Lippert, Bernhard, J. Am. Chem. Soc. 1998, 120 , 12000-12007.
The X-ray crystal structures of two complexes of the composition trans-[Pt(NH3)2(9-EtG-N7)(1-MeC-N3‘)]X·nH2O (1) with 9-EtG = 9-ethylguaninate and 1-MeC = 1-methylcytosine are reported. 1b (X = picrate, n = 1) crystallizes to produce a dimetalated base quartet, held together by H-bonding interactions between pairs of cations. This feature essentially corresponds to the solution structure previously proposed by us on the basis of 1H NMR and ESI-MS, with a H-bonding interaction between the aromatic H5‘ proton of 1-MeC and the deprotonated N1 position of 9-EtG. 1c (X = trifluoromethanesulfonate, n = 0) crystallizes in a radically different fashion as a consequence of nucleobase rotation about the Pt−N bond, leading to a reversed Hoogsteen arrangement without any intracomplex H bonding between the two bases. In the solid-state structure of 1b short intermolecular H bonds exist between the exocyclic NH2 group of 1-MeC and O6 of 9-EtG (2.715(7) Å). Considerably longer intra- (N4‘(1-MeC)···O6(9-EtG), 3.229(6) Å) and intermolecular (C5‘(1-MeC)···N1(9-EtG), 3.548(7) Å) H bonds are primarily a consequence of considerable base twisting, presumably caused by stacking between the guanine residues and the picrate anions. In DMSO-d6 solution, the cyclic base quartet structure is favored, regardless of the nature of the anion X (X = picrate, trifluoromethanesulfonate, perchlorate, nitrate). An association constant KD = 44.1 ± 3.2 M-1 for the dimerization has been determined.
If Watson–Crick and Hoogsteen sites of guanine are blocked, hydrogen bonding with cytosine is via N2 and N3
Sigel, Roland K. O.; Freisinger, Eva; Lippert, Bernhard, Chem. Commun. 1998, 0 , 219-220.
A novel hydrogen bonding scheme between cytosine and guanine nucleobases is observed if both N1 and N7 positions of the guanine are blocked by a metal entity and a methyl group, respectively.
1997
Stabilities and Structures of Metal Ion Complexes of Adenosine 5‘- O -Thiomonophosphate (AMPS 2- ) in Comparison with Those of Its Parent Nucleotide (AMP 2- ) in Aqueous Solution
Sigel, Roland K. O.; Song, Bin; Sigel, Helmut, J. Am. Chem. Soc. 1997, 119 , 744-755.
The stability constants of the 1:1 complexes formed between Mg2+, Ca2+, Ba2+, Mn2+, Co2+, Ni2+, Zn2+, or Cd2+ and AMPS2-, i.e., of the M(AMPS) complexes, were determined by potentiometric pH titrations (25 °C; I = 0.1 M, NaNO3). For the Mn2+/AMPS, Co2+/AMPS, Ni2+/AMPS, and Cd2+/AMPS systems also the protonated species M(H;AMPS)+ were quantified, and for the Zn2+/AMPS system, the stability of the hydroxo species Zn(AMPS)(OH)-, which results from the Zn2+−thio coordination, could be determined. On the basis of previously established log versus p straight-line plots (R-MP2- = simple monophosphate ester ligands without further coordinating groups; Sigel, H.; et al. Helv. Chim. Acta 1992, 75, 2634), it is concluded that the alkaline earth ions in the M(AMPS) complexes are coordinated to the thiophosphate group with the same intensity as to a normal phosphate group. For the M(AMPS) complexes of Mn2+, Co2+, Ni2+, Zn2+, and Cd2+, it is shown by comparison with the corresponding M(AMP) complexes and by employing the mentioned straight-line plots that the stability increase is larger than may be expected due to macrochelate formation, which means that the metal ions also bind to the sulfur atom of the thiophosphate group. The stability increases amount for Mn(AMPS), Zn(AMPS), and Cd(AMPS) to about 0.2, 0.7, and 2.4 log units, respectively, and the estimated approximate percentages of the sulfur-coordinated species are about 30, 80, and 100{\%}, respectively. Furthermore, comparisons between these stability increases and the solubility products for the corresponding metal ion sulfides, MIIS, as well as with the stability increases due to the M2+−thioether interaction observed for the complexes of tetrahydrothiophene-2-carboxylate, which also result in straight-line plots, further support the conclusions about metal ion−sulfur binding in the mentioned M(AMPS) complexes. The indicated correlations allow also an estimate for the extent of the M2+−sulfur interaction in Pb(AMPS) and Cu(AMPS). The various isomers of the M(H;AMPS)+ species are analyzed in a microconstant scheme, and estimations about their formation degrees are presented; for example, for the Cd2+ system, (H·AMPS·Cd)+ is the dominating isomer, which has the proton at N1 and Cd2+ at the thiophosphate group. It is evident that for metal ions like (Mn2+), Zn2+, or Cd2+ the metal ion binding properties of the parent compound AMP2- and its thio analogue AMPS2- differ considerably, and therefore, great care should be exercised in enzymatic studies where AMPS2- is employed as a probe for AMP2- in the presence of metal ions. Regarding studies of ribozymes, it is of interest that plots are presented (pseudo-first-order rate constants versus complex stabilities) which suggest that on top of a sulfur−metal ion interaction during the transition state of the rate-determining step of the hydrolytic cleavage of an oligonucleotide containing a bridged internucleotide 5‘-phosphorothioate RNA linkage also an oxygen−metal ion interaction occurs and that the two effects are “additive”.
Acid-Base Properties of Adenosine 5′-O-Thiomonophosphate in Aqueous Solution
Song, Bin; Sigel, Roland K. O.; Sigel, Helmut, Chem. Eur. J. 1997, 3 , 29-33.
The acidity constants of H2-(AMPS)± were determined by potentiometric pH titrations in aqueous solution at 25°C and I=0.1M (NaNO3). Titrations with a combined single-junction glass electrode were hampered in the presence of AMPS by a “poisoning” effect; the problem could be avoided by use of two separated electrodes. The values of the acidity constants PKHH2(AMPS) = 3.72 ± 0.03 and pKHH2(AMPS) = 4.83 ± 0.02 are relatively close to each other; the buffer regions of the two equilibria overlap, and therefore a micro acidity constant scheme was developed and the constants for the various sites calculated. It is concluded that the thiophosphateprotonated species (AMPS⋅H)− dominates at about 75{\%} occurrence, while the form (H⋅AMPS)−, with the proton at the N1 site of the adenine residue, occurs at about 25{\%}. Semiempirical AM1 and PM3 calculations including water as a solvent locate the proton in (AMPS⋅H)− mainly on the terminal oxygen atoms rather than the sulfur. The acid-base properties of H2(AMPS)± are considerably more complicated than those of the parent nucleotide, H2(AMP)±; for the latter the two (intrinsic) acidity constants are well separated and consequently practically all protons have left the N1 site before deprotonation at the monoprotonated phosphate group occurs. Finally, an estimate for the acidity constants of H2(ATPγS)2- is given.