
 Inhalt Index
Inhalt IndexEine C=C Doppelbindung ist die funktionelle Gruppe, die für die Alkene (Olefine) charakteristisch ist. Sie haben weniger H-Atome pro C-Atom als Alkane, und werden deshalb als ungesättigt bezeichnet, z.B.(Trivialnamen !):
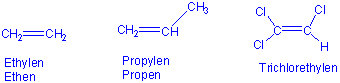
In der IUPAC-Nomenklatur wird die einfachere Endung -en anstatt -ylen benützt. Kompliziertere Systeme erfordern Anpassungen und Erweiterungen der Nomenklaturregeln für Alkene:
i) Finden Sie die längste Kohlenstoff-Kette, die beide an der Doppelbindung beteiligte C-Atome enthält:
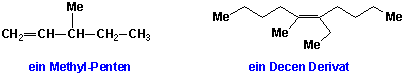
ii) Die Nummerierung beginnt an dem Ende, das der Doppelbindung näher ist. In Cycloalkenen sind die Kohlenstoffatome 1 und 2 immer die, zwischen denen die Doppelbindung liegt:
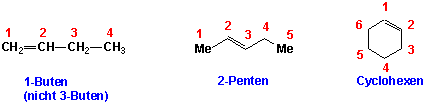
iii) Die Substituenten und ihre Positionen werden dem Namen des Alkens als Präfixe vorgestellt:
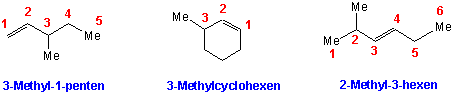
iv) Zwei Substituenten können auf denselben oder auf entgegengesetzten Seiten der Doppelbindung stehen. Die erste stereochemische Anordung wird cis genannt, die zweite trans :
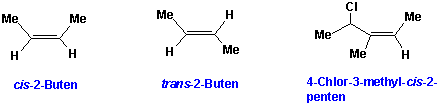
Ein alternatives und allgemeineres System zur Benennung von cis/trans-Isomeren wurde von der IUPAC eingeführt: Das E,Z-System (später).
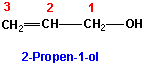
v) Wenn andere funktionelle Gruppen vorhanden sind, können sie gegenüber der Doppelbindung Vorrang haben z.B Alkenole :
vi) Substituenten, die eine Doppelbindung enthalten, werden Alkenyl genannt:
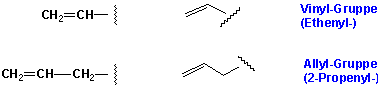
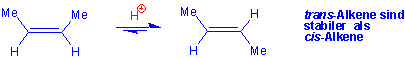
Die Rotation um eine C=C Doppelbindung benötigt einen hohen Energiebetrag, weil die π-Bindung (π-Bindungsdissoziationsenergie 272 KJ/mol) gebrochen werden muss:
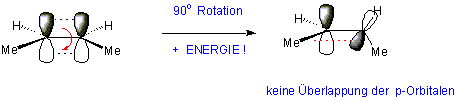
Normalerweise sind unter 300oC die meisten Doppelbindungen konfigurationsstabil. Aber mit einer starken Säure kann die Isomerisierung katalysiert werden, und beim Gleichgewicht steht das trans-Isomer im Überschuss. Die trans-Isomere sind stabiler als die cis-Isomere.
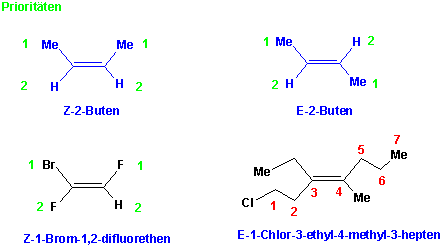
Um bei den mehrfach substituierten Alkenen verschiedene Isomeren eindeutig kennzeichnen zu können, benutzt man die Sequenzregel von Cahn, Ingold und Prelog. Nach diesen Regeln sind alle möglichen Substituenten in einer Reihe nach abnehmender Priorität geordnet.
Die Priorität der Substituenten ergibt sich aus der Abnahme der Ordungszahl, der direkt an die Doppelbindung gebundenen Atome. z.B. :
35Br > 17Cl > 8O > 7N > 6C > 1H usw.
Dasjenige Isomer, bei welchem die beiden Gruppen höherer Priorität auf derselben Seite der Doppelbindung liegen, wird als Z-Isomer, das andere als E-Isomer bezeichnet, z.B.:
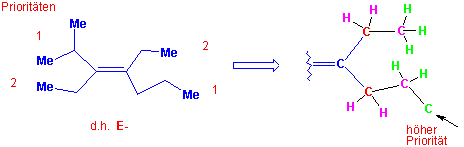
Sind zwei oder mehr mit der Doppelbindung verknüpfte Atome identisch, so bestimmt die Atomnummer des benachbarten Atoms die Prioritätenfolge :
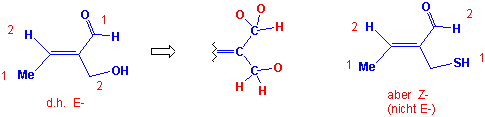
Doppelt gebundene Atome sind als Äquivalent zur gleichen Anzahl einfach gebundener Atome zu betrachten, z. B. -CH=O hat ein C-Atom mit einer Doppelbindung zum O-Atom und wird als Äquivalent zu einem C-Atom mit zwei einfach gebundenen O-Atomen betrachtet:
Da verschiedene funktionelle Gruppen aber auch gleichartige chemische Reaktionen zeigen, ist es möglich, das Gesamtgebiet der Organischen Chemie nach Reaktionstypen zu gliedern. Betrachtet man nur die Veränderungen des Molekülskelettes und lässt den genauen Ablauf unberücksichtigt, so kann man vier Haupttypen von Reaktionen unterscheiden :
Substitutionen, Additionen, Eliminierungen und Umlagerungen
In chemischen Reaktionen werden Kovalenzbindungen gebrochen und gebildet. Kovalenzbindungen können auf zwei Arten gebrochen werden:
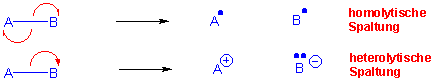
Bei der homolytischen Bindungstrennung entstehen Atome und/oder Radikale, während bei der heterolytischen Bindungstrennung Ionen, eventuell auch neutrale Molekule gebildet werden können. Umgekehrt, kann bei der Bindungsneubildung die Kovalenzbindung durch einen radikalischen oder durch einen polaren Prozess gebildet werden :
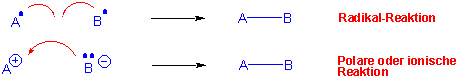
In polaren Reaktionen werden die Elektron-Bewegungen durch Pfeile gekennzeichnet, in dem ein Nucleophil ("nucleus-loving", ein elektronreiches Atom oder Atomgruppe) seine Elektronen auf ein Elektrophil ("electron-loving", ein elektronarmes Atom oder Atomgruppe) überträgt, um eine neue Kovalenzbindung zu bilden :
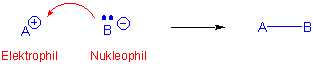
Wie unser Orbitalbild zeigt, besteht die Doppelbindung aus zwei unterschiedlichen Bindungen : einer σ- und einer π-Bindung :
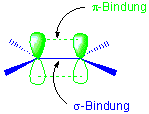
Chemische Bindungen entstehen durch die Überlappung von Orbitalen, und die Bindungsstärke wird von der Effektivität dieser Überlappung bestimmt. Wegen der räumlichen Anordung der Orbitale ist zu erwarten, dass die Orbital-Überlappung in einer σ-Bindung besser ist als in einer π-Bindung. Weil die Überlappung in π-Orbitalen geringer ist als in σ-Orbitalen, sollte die Abspaltung eines Elektrons aus einem Alken aus dem energetisch höher liegenden π-Niveau erfolgen, weil π-Elektronen weniger stark gebunden sind als σ-Elektronen.
Eine C=C Doppelbindung (und ebenso eine Dreifachbindung) stellt ein Zentrum von relativ hoher negativer Ladungsdichte dar, und es ist deshalb verständlich, dass sie besonders leicht von elektrophilen Reagentien angreifbar ist: d.h. C=C Doppelbindungen sollten sich als Nucleophile verhalten, z.B. Addition von HCl :
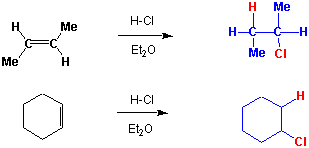
Mechanismus der Addition :
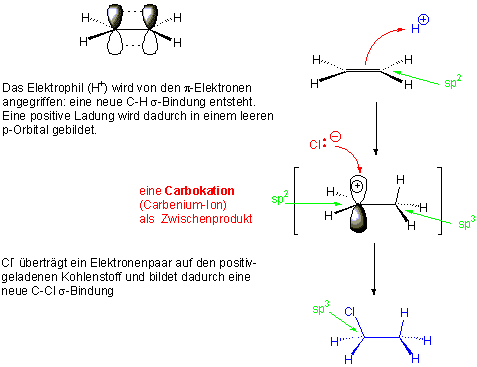
Ein Carbokation (Carbeniumion) entsteht als Zwischenprodukt. Zwischenprodukte treten oft in mehrstufigen Reaktionen auf und sind gewöhnlich instabil und reagieren rasch weiter.
Die chemische Thermodynamik befasst sich mit den Energieänderungen in Reaktanten und Produkten bei chemischen Reaktionen. Diese sind ein Mass dafür, wo ein chemisches Gleichgewicht liegt :
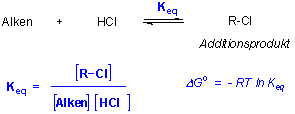
Keq gross : Gleichgewicht liegt auf der rechten Seite : Das Energieniveau der Produkte ist niedriger als das der Reaktanten (exotherme Reaktion)
Keq klein (<1) Gleichgewicht liegt auf der linken Seite : Das Energieniveau der Produkte ist höher als das der Reaktanten (endotherme Reaktion).
Die Gleichgewichtslage wird durch ΔGo, die Differenz zwischen den freien Enthalpien der Produkte und der Reaktanten, bestimmt.
z.B. für A --> B
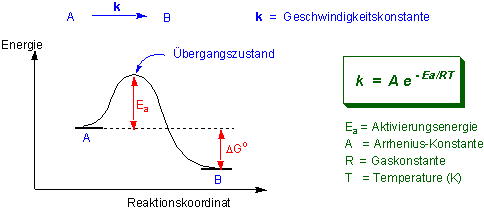
Die Energie, die zur Überschreitung der Energiebarriere nötig ist, erlangt ein Molekül normalerweise durch Zusammenstösse mit anderen Molekülen. Die Häufigkeitsverteilung der kinetischen Energie bei einer gegebenen Temp. wird durch die Boltzmann-Verteilung angegeben. Bei RT beträgt die mittlere kinetische Energie der Moleküle einer organischen Verbindung nur etwa 2.5 KJ/mol. Viele organische Reaktionen besitzen Aktivierungsenergien von 40-150 KJ/mol. Damit eine Reaktion bei Raumtemperatur abläuft, kann die Aktivierungsenergie bis auf etwa 80 KJ/mol liegen. Reaktionen mit höherer Aktivierungsenergie finden nur bei höherern Temperaturen statt.
Für die elektrophile Addition von HCl an Alkene :
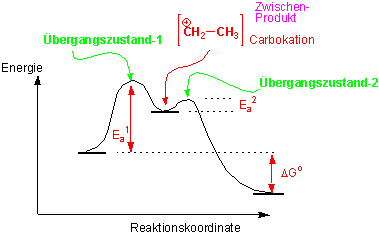
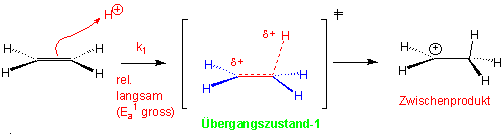
Die Strukturen, die auf Energiemaxima liegen, heissen Übergangszustände. Sie können weder isoliert noch beobachtet werden. Im ersten Übergangszustand ist die C=C Doppelbindung partiell gespalten und die neue C-H σ-Bindung partiell gebildet:
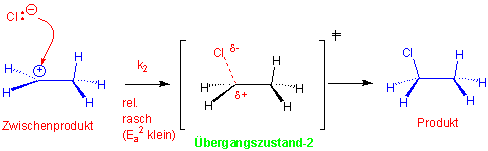
Das Zwischenprodukt, in diesem Fall ein Carbokation, liegt in einem Energieminimum, ist aber instabil weil es auf einem hohen Energieniveau liegt. Der zweite Übergangszustand benötigt nur einen sehr kleinen Energiebetrag und läuft deswegen sehr rasch ab:
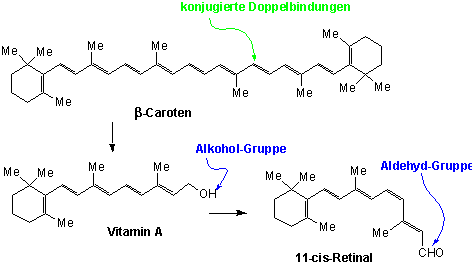
Karotten stellen eine reiche Quelle von β-Caroten dar, was für die körperliche Bildung von Vitamin A im Körper verwendet werden kann. β-Caroten wird in der Leber durch Enzym-katalysierte Reaktionen in Vitamin A und 11-cis-Retinal umgewandelt :
In unseren Augen findet man zwei besondere Zell Typen: Stäbchen und Zapfen (rod cells und cone cells). In den "Rod-cells" wird 11-cis-Retinal zum Protein Opsin gekuppelt, und dadurch wird Rhodopsin gebildet. Rhodopsin ist ein integrales Membranprotein und ist der Hauptphotorezeptor unseres Sehvermögens. 1958 zeigten George Wald und Mitarbeiter, das Licht das 11-cis-Retinal des Rhodopsins zu all-trans-Retinal isomerisiert. Diese Isomerisierung, das Primärereignis des Sehvorgangs, ändert die Geometrie des Retinals und dadurch die Konformation des Opsin Moleküle wesentlich. Dadurch ist ein Photon in atomare Bewegung umgewandelt worden:
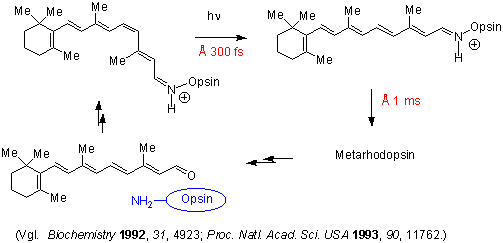
Danach wird all-trans-Retinal (durch eine Hydrolyse) freigesetzt, und in einer Dunkelreaktion zu 11-cis-Retinal isomerisiert.
![]()
