
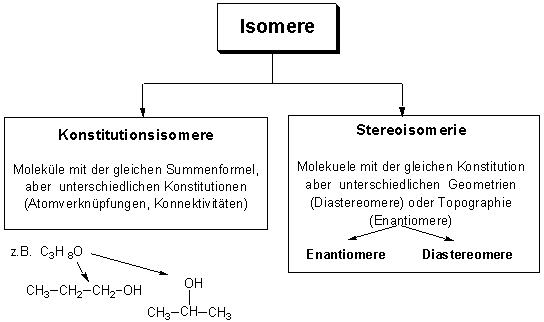
 Inhalt Index
Inhalt IndexIn den vorhergehenden Kapiteln haben wir zwei Arten von Isomerie kennengelernt (siehe Kapitel 2.2, 2.8, 3.2 ), die Konstitutionsisomerie, und die Stereoisomerie :
Die Stereochemie befasst sich mit der Geometrie (oder Struktur) und Händigkeit (oder Topographie) von Molekülen, ihren dynamischen Aspekten, und die Verhältnisse zwischen diesen und der Reaktivität des Moleküls.
Wir werden hier den Begriff Struktur wie folgt definieren : die Art und relative Position von Atomen im drei-dimensionalen Raum, die ein stabiles Molekularsystem darstellt, heisst seine Struktur. Wir haben oftmals durch eine Röntgenstrukturanalyse Einblick in die Struktur eines Moleküls. Mit deren Hilfe lässt sich die Struktur eines Moleküls im kristallinen Zustand aufklären. Nach der Bestimmung der Positionen der Atome kann man Bindungslängen und -winkel sowie andere geometrische Faktoren ableiten.
Beispielen von Stereoisomeren, die uns schon begegnet sind :
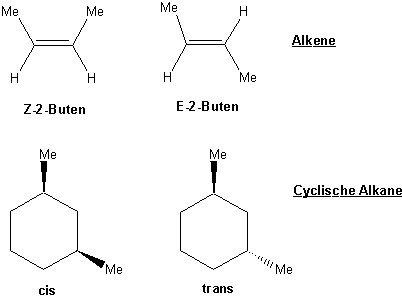
In diesem Kapitel stellen wir noch einen Typ von Stereoisomeren vor, der sich aus der "Händigkeit" bestimmter Moleküle ergibt. Wir werden sehen, dass es Strukturen gibt, die sich nicht mit ihrem Spiegelbild zur Deckung bringen lassen, genauso wie Ihre linke Hand nicht deckungsgleich mit Ihrer rechten ist. Beide Strukturen sind daher verschiedene Objekte, sie können unterschiedliche Eigenschaften besitzen und auf andere Weise reagieren.
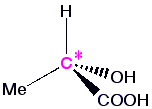
Betrachten wir das Molekül Milchsäure (Lactic acid), das in zwei Formen existieren kann :
 | 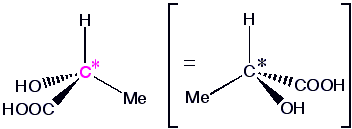 |
| Spiegelebene | |
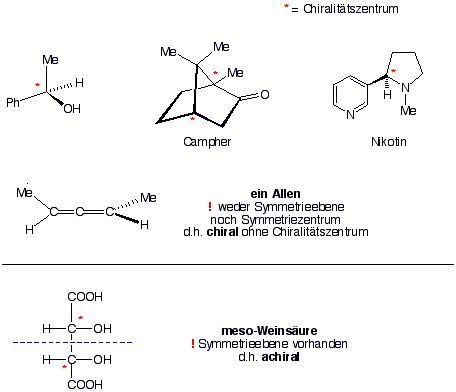
Beide Moleküle stehen zueinander wie Objekt und Spiegelbild und lassen sich nicht zur Deckung bringen. Wollte man eines in das andere überführen, müsste man Bindungen aufbrechen und neu knüpfen. Paare von Molekülen, die sich zueinander spiegelbildlich verhalten, aber nicht deckungsgleich sind, bezeichnet man als Enantiomere. Moleküle, die in zwei enantiomeren Formen vorkommen können, sind chiral.
Das wichtigste Kriterium für Chiralität ist, dass sich Objekt und Spiegelbild nicht zur Deckung bringen lassen, obwohl sie die identische Konnektivität besitzen.
Weitere Beispiele:
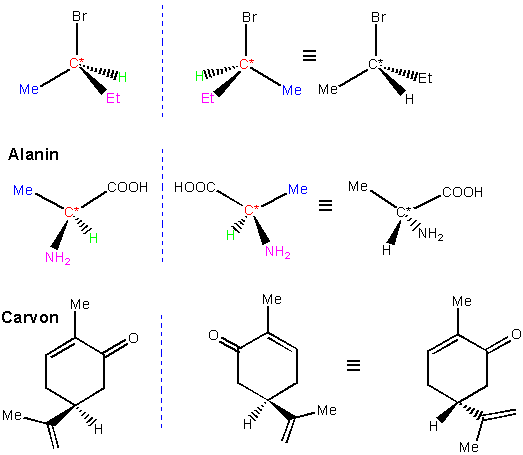
 In den obigen Beispielen, enthalten alle gezeigten Moleküle ein Atom, an das vier verschiedene Substituenten gebunden sind. Man bezeichnet es als asymmetrisches Atom oder Chiralitätszentrum.
In den obigen Beispielen, enthalten alle gezeigten Moleküle ein Atom, an das vier verschiedene Substituenten gebunden sind. Man bezeichnet es als asymmetrisches Atom oder Chiralitätszentrum.
Aber viele andere chirale Moleküle haben kein Chiralitätszentrum.
Wie lassen sich chirale Strukturen von achiralen unterscheiden ?
Es ist nicht immer einfach zu sehen, ob ein Molekül chiral ist oder nicht. Absolut narrensicher ist es, Modelle des Moleküls und seines Spiegelbildes zu bauen und zu sehen, ob man sie zur Deckung bringen kann. Leider erfordet dieses Verfahren manchmal viel Zeit. Aber es gibt zwei Hilfen, mit denen man schnell feststellen kann, ob ein Molekül chiral ist oder nicht. Sie basieren auf den Symmetrieeigenschaften des Moleküls.
Durch bestimmte Symmetrieoperationen, die man am Molekül vornehmen kann, bleiben dessen Struktur und die Position der Atome im Raum unverändert. Wir brauchen nur zwei zu berücksichtigen :
die Einführung einer Symmetrieebene oder eines Symmetriezentrums.
Eine Symmetrieebene schneidet das Molekül derart, dass der Teil der Struktur, der auf der einen Seite der Ebene liegt, das Spiegelbild des Teils auf der anderen Seite ist, z.B.:
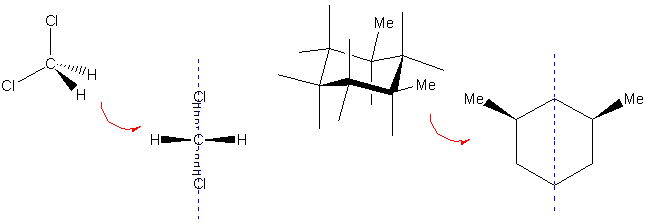
Ein Symmetriezentrum ist ein Punkt in einem Molekül, der jede Gerade, die durch ihn gezeichnet wird, in zwei gleichgrosse Gruppen von Punkten auf jeder Seite mit derselben Umgebung teilt. Es kann immer nur einen solchen Punkt geben, z.B.:
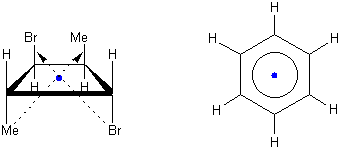
Um ein chirales Molekül von einem achiralen zu unterscheiden, müssen wir uns nur merken, dass chirale Moleküle weder ein Symmetriezentrum noch eine Symmetrieebene enthalten dürfen. Liegt eines von beiden Symmetrieelementen im Molekül vor, ist es achiral.
Noch weiter verallgemeinert: Moleküle mit einer Symmetrieebene oder einem Symmetriezentrum besitzen zwei Arten von sogennanten Drehspiegelachsen (Sn). Drehspiegelachsen bestehen aus einer Drehachse (Cn) und einer Spiegelebene senkrecht zur Drehachse. Chirale Moleküle dürfen eine Drehachse besitzen aber nicht Drehspiegelachsen (Sn), wobei eine Symmetrieebene=S1 und ein Symmetriezentrum=S2 gleich sind.
Andere chirale Moleküle :
Ein 1 : 1-Gemisch von Enantiomeren bezeichnet man als Racemat oder racemisches Gemisch
Wird ein Enantiomer über irgendeinen Prozess mit seinem Spiegelbild ins Gleichgewicht gebracht, spricht man von Racemisierungi.
Z.B. Ein Racemisierung kann durch eine Konformationsänderung erfolgen:

Da es sich hier nur um eine Konformationsänderung handelt, die nur wenig Energie braucht, können die beiden enantiomeren Konformationen nicht getrennt und in enantiomeren-reiner Form erhalten werden. Bernsteinsäure muss deshalb als ein achirales Molekül betrachtet werden.
Dagegen, z.B.:
Bei Atropisomeren handelt es sich um Rotamere, bei denen die Rotation um eine kovalente Einfachbindung durch sterisch anspruchsvolle Substituenten derart eingeschränkt ist, dass Konformere isoliert werden können.
Fassen wir zusammen ; Chirale Moleküle sind Stereoisomere, bei denen Bild- und Spiegelbild nicht deckungsgleich sind. Man bezeichnet die beiden Isomere als Enantiomere. In vielen chiralen organischen Molekülen ist ein Chiralitätszentrum enthalten, in anderen nicht. Ein chirales Molekül besitzt weder eine Symmetrieebene noch ein Symmetriezentrum (im allgemeinen - keine Sn-Symmetrie).
Wie können wir nun bestimmen, welches Enantiomer welches ist ? Und wenn wir die Antwort wissen, gibt es eine Möglichkeit, wie man ein Enantiomer eindeutig benennen und es von seinem Spiegelbild unterscheiden kann ?
Enantiomere Moleküle sind sehr ähnlich, weil sie identische Strukturen haben ; d.h. identische Bindungslängen, Bindungswinkel, und deswegen denselben Energieinhalt. Aus diesem Grund haben Enantiomere identische physikalische (Schmelzpunkt, Siedepunkt usw.) und chemische Eigenschaften, mit Ausnahme ihrer Wechselwirkung zu anderen chiralen Molekülen und Objekten. Sie können auch nicht durch normale Verfahren (wie z.B. durch Kristallisation, fraktionierte Destillation, verschiedene chromatographische Methoden) getrennt werden
Schickt man jedoch einen Strahl von linear polarisierten Licht durch eine Probe eines der beiden Enantiomere, wird die Schwingungsebene des einfallenden Lichtes um einen bestimmten Betrag in eine Richtung gedreht (entweder im oder gegen den Uhrzeigersinn). Wiederholt man dasselbe Experiment mit dem anderen Enantiomer, wird die Schwingungsebene um genau denselben Betrag, nur in die andere Richtung, gedreht.
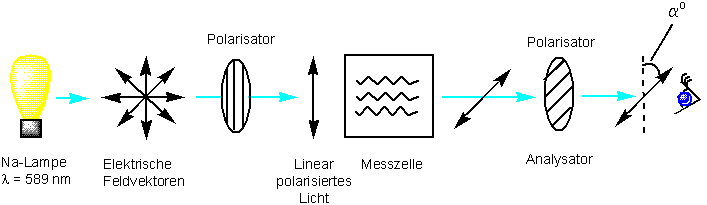
Ein Enantiomer, das die Ebene des polarisierten Lichts im Uhrzeigersinn dreht, bezeichnet man als rechtsdrehend (dextrorotatory) und nennt es willkürlich (+)-Enantiomer. Entsprechend ist das andere Enantiomer, das die Ebene gegen den Uhrzeigersinn dreht, das linksdrehende (levorotatory) oder das (-)-Enantiomer.
Enthält das Polarimeter achirale Moleküle, bleibt die Richtung unverändert, die Probe ist optisch inaktiv.
Die gemessene Drehung ist eine makroskopische Eigenschaft - die Summe über alle Rotationen durch die einzelnen Moleküle. Man bezeichnet dies als optische Drehung, und eine Probe, die Anlass zu einer optischen Drehung gibt, als optisch aktiv.
Der Wert der beobachteter optischer Drehung hängt von der Konzentration und Struktur des optisch aktiven Moleküls ab, der Länge der Messzelle, der Wellenlänge des Lichtes, des Lösungsmittel und der Temperatur. Deswegen wird eine spezifische Drehung ausgerechnet :
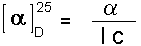 |
[α]D25 = spezifische Drehung bei 589 nm und 25oC.
α = beobachtete Drehung l = Länge der Messzelle in dm (1 dm = 10 cm) c = Konzentration in g/ml |
Die spezifische Drehung einer optisch aktiven Verbindung ist eine physikalische Konstante, die für diese Substanz charakteristisch ist, ebenso wie der Schmelzpunkt, der Siedepunkt und die Dichte.
Beispiel:
Campher (1.5 g) wird in 10 ml Chloroform (CHCl3) gelöst. In einer 10 cm Küvette wird eine Drehung von +6.645 gemessen. Berechnen Sie den spezifischen Drehwert für Campher.

Das (-)Enantiomer von Campher dreht also die Ebene gegen den Uhrzeigersinn mit einer spezifischenDrehung von -44.3o, während das (+)Enantiomer die Ebene um +44.3o im Uhrzeigersinn dreht.
Daraus folgt, dass die optische Drehung eines Racemats null ist. Es ist optisch inaktiv.
Um an einem Enantiomerengemisch optische Aktivität beobachten zu können, muss eines von beiden Enantiomeren im Überschuss vorliegen. Mit Hilfe des Wertes der spezifischen Drehung können wir die Zusammensetzung von Gemischen zweier Enantiomerer berechnen :
z.B.
optische Reinheit = ([α]D-beobachtet / [α]D x 100%
Wie können wir die Molekülstruktur eines reinen Enantiomers einer chiralen Verbindung ermitteln ?
Z.B. Milchsäure :
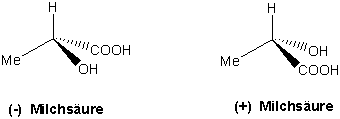
Moleküle wie (+)- und (-)-Milchsäure, die sich zueinander spiegelbildlich verhalten (d.h. Enantiomere), unterscheiden sich in ihren Topographien (aber nicht in ihrer Strukturen. Sie haben identische Strukturen). Sie haben verschiedene räumliche Anordnungen der Substituenten um das Chiralitätszentrum, d.h. sie haben unterschiedliche Händigkeiten, oder absolute Konfigurationen.
Ist es möglich, die absolute Konfiguration eines Enantiomers durch Messen des spezifischen Drehwerts zu bestimmen? Leider nicht. Es besteht keine eindeutige Beziehung zwischen dem Vorzeichen des Drehwerts und der absoluten Konfiguration.
Um Enantiomere eindeutig zu benennen, brauchen wir ein System, mit dessen Hilfe wir die Händigkeit des Moleküls angeben können. Ein solches System ist von Cahn, Ingold (London) und Prelog (ETH Zürich) entwickelt worden (CIP-Nomenklatursystem).
Wir werden uns im folgenden auf die Regeln beschränken, die für Stereoisomerie mit asymmetrischen C-Atomen entwickelt wurden:
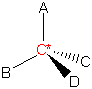
1) alle vier Substituenten nach abnehmender Priorität (siehe Kapitel 3.2) als 1, 2, 3, und 4 zuordnen (d.h. nach abnehmender Ordnungszahl):
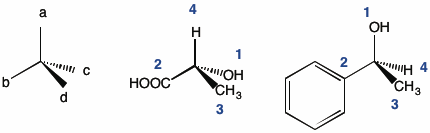
2) als nächstes dreht man das Molekül so, dass der Substituent mit der geringsten Priorität am weitesten vom Betrachter entfernt ist:
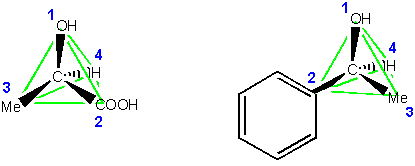
3) Bewegt man sich, um von 1 über 2 nach 3 zu gelangen, gegen dem Uhrzeigersinn, besitzt das Chiralitätszentrum die absolute Konfiguration S (sinister, lateinisch, links). Bewegt man sich im anderen Fall im Uhrzeigersinn, ist die absolute Konfiguration R (rectus, lateinisch, rechts).
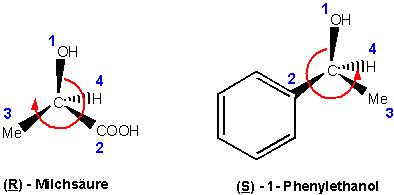
Das Symbol R oder S wird in Klammern vor den Namen der chiralen Verbindung gesetzt.
Weitere Beispiele :
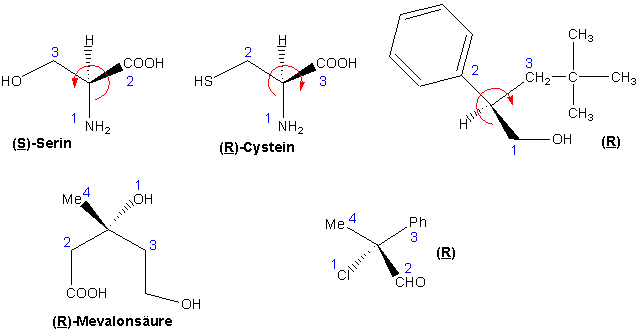
Aus welchen Gründen könnten wir dann einem Stereoisomeren einer chiralen Verbindung ein positives, dem anderen ein negatives Vorzeichen von [α] zuordnen ? Diese Zuordnung ist auf dem direktesten Weg durch eine bestimmte Form der Röntgenstrukturanalyse möglich, die anomale Dispersion, die nicht nur die Struktur sondern auch die absolute Konfiguration ermittelt. Die absolute Konfiguration eines Enantiomers lässt sich auch durch chemische Korrelation mit einer Struktur, deren eigene absolute Konfiguration durch Röntgenstrukturanalyse ermittelt wurde, ableiten.
Vor der Entdeckung der Röntgenstrukturanalyse war die absolute Konfiguration chiraler Moleküle unbekannt. Um wenigstens ein einheitliches System von relativen Konfigurationen zu schaffen, wurden willkürlich beiden Enantiomeren von 2,3 Dihydroxypropanal (Glycerinaldehyd), einer synthetisch wichtigen Substanz, die sich in eine Fülle anderer chiraler Moleküle überführen lässt, Konfigurationen zugeordnet.
Man postulierte, dass das rechtsdrehende Enantiomer eine Struktur besässe, die mit D-Glycerinaldehyd bezeichnet wurde. Die Bezeichnung "D" leitet sich ursprünglich vom lateinischen Wort "dexter" für rechts ab, bezieht sich aber nicht auf das Vorzeichen der Drehung des linear polarisierten Lichts, sondern auf die relative Anordnung der Substituenten, die willkürlich, wie unten gezeigt, geschrieben wurde.
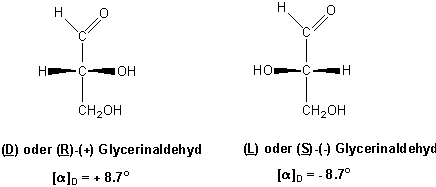
Entsprechend bezeichnete man das linksdrehende Isomer als L-Glycerinaldehyd (L von lateinisch: laevulus, links). Damit die Buchstaben D und L, die, wie wir noch einmal betonen möchten, die Konfiguration, und nicht den Drehsinn angeben nicht mit diesem verwelchselt werden, schreibt man das Vorzeichen des Drehsinns (+ oder -) in Klammern hinter die Angabe der Konfiguration und vor den Namen der Verbindung. Allen chiralen Verbindungen, die durch chemische Prozesse mit D-(+)-Glycerinaldehyd korreliert werden konnten- das bedeutet, dass man sie mit Hilfe von chemischen Reaktionen, die die Konfiguration am chiralen Kohlenstoff nicht verändern, in rechtsdrehenden Glycerinaldehyd überführen konnte - wurde die D-Konfiguration zugeordnet, ihren Spiegelbildisomeren die L-Konfiguration. Beispiele für Moleküle mit D- und L-Konfiguration sind unten abgebildet. Erst im Jahre 1951 gelang es mit Hilfe der Röntgenstrukturanalyse, die absolute Konfiguration des D-Glycerinaldehyds zu bestimmen.
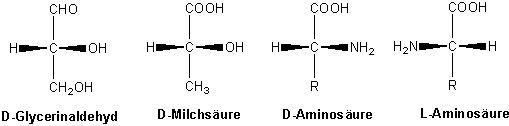
Die D.L.-Nomenklatur wird noch stets bei den Zuckern und den Aminosäuren verwendet, aber sollte durch die R/S-Nomenklatur ersetzt werden.
Eine Fischer-Projektion (nach Emil Fischer, 1852-1919) ist eine Standardmethode zur zweidimensionalen Abbildung tetraedrischer Kohlenstoffatome und ihrer Substituenten. In dieser Darstellungsmethode wird das Molekül als Kreuz mit dem chiralen Kohlenstoff im Schnittpunkt der beiden Achsen gezeichnet. Die waagerechten Linien stellen Bindungen dar, die auf den Betrachter zu gerichtet sind, senkrechte Linien weisen von ihm weg :
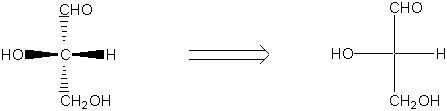
In Molekülen mit mehreren Chiralitätszentren :
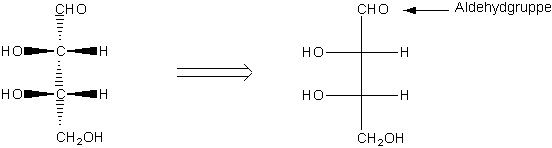
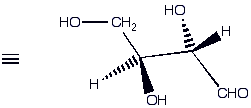
| Beide räumliche Anordnungen lassen sich durch einfache Drehung um die zentrale C-C-Bindung ineinander überführen. |
||
| Fischer Projektion (räumliche Darstellung mit ekliptischer Anordnung) | stabile gestaffelte Anordnung |
In Molekülen, die mehrere Chiralitätszentren (Asymmetrische C-Atome) besitzen, wird es komplizierter. Jetzt sind mehrere Stereoisomere möglich. Als Beispiel werden wir zuerst die Aminosäure Threonin betrachten. Threonin besitzt zwei Chiralitätszentren, und jedes davon kann entweder R oder S konfiguriert sein :
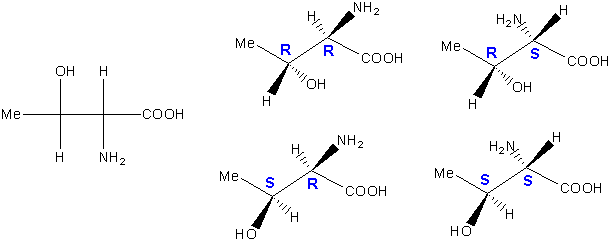
Durch eine einfache Permutation kann man feststellen, dass hier vier Stereoisomere möglich sein sollten. Die vier Stereoisomere können auch mit Hilfe der Fischer-Projektionen geschrieben werden :
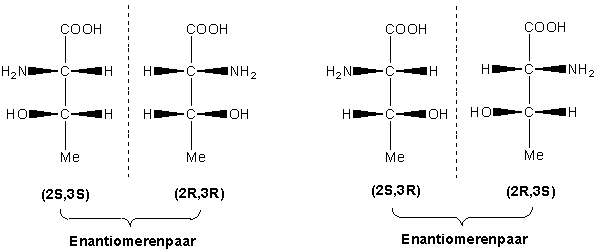
Jetzt lautet eine wichtige Frage: wie verhalten sich die Stereoisomeren zueinander ? Als Bild zum Spiegelbild, oder nicht ? Das ist ziemlich leicht festzustellen. Wenn wir die Moleküle genauer betrachten, sehen wir, dass wir zwei Paare von Verbindungen haben : ein R,R / S,S-Paar, und ein R,S / S,R-Paar. Das R,R Isomer ist das Spiegelbild des S,S-Isomers. Und das R,S-Isomer ist das Spiegelbild des S,R-Isomers. Wir haben also zwei Enantiomerenpaare.
Stereoisomere, die sich nicht wie Bild und Spiegelbild verhalten, sind jedoch Diastereomere.
Wenn wir nochmals ein Diastereomerenpaar betrachten, können wir noch etwas wichtiges feststellen:
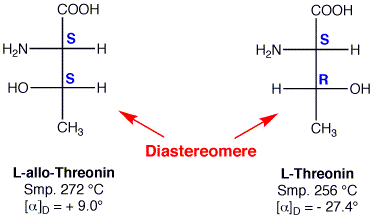
Diastereomere haben unterschiedliche Strukturen (oder Geometrien). D.h., wenn wir jede entsprechende Bindungslänge, Bindungswinkel und Torsionswinkel betrachten können wir feststellen dass sich in mindestens einem dieser Werte die Diastereomeren unterscheiden. Das bedeutet auch, dass Diastereomere unterschiedliche Energieinhalte haben sollten, was weiter bedeutet, dass im Gegensatz zu Enantiomeren, diastereomere Moleküle unterschiedliche physikalische und chemische Eigenschaften haben müssen.
Sie lassen sich z.B. durch fraktionierende Destillation bzw. Kristallisation oder durch chromatographische Methoden trennen. Sie unterscheiden sich in ihrem Schmelz- und Siedepunkt und in ihrer Dichte, genau wie Konstitutionsisomere, und zeigen verschiedene spezifische Drehwerte.
Dieselbe stereochemische Beziehung gilt auch für Systeme, bei denen derartige Zentren durch ein oder mehrere Atome getrennt sind.
z.B.:

Wieviele Stereoisomere kann man erwarten, wenn beide Zentren gleich substituiert sind ?
z.B. Weinsäure (Tartaric acid) :
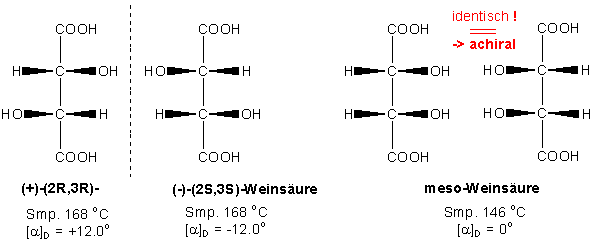
| Beide räumliche Anordnungen lassen sich durch einfache Drehung um die zentrale C-C-Bindung ineinander überführen. |
||
| Fischer Projektion (räumliche Darstellung mit ekliptischer Anordnung) | stabile gestaffelte Anordnung |
Beim ersten Paar von Stereoisomeren, die R,R- und S,S-Stereoisomeren, lässt sich klar erkennen, dass es sich um einen Enantiomerenpaar handelt. Betrachtet man das zweite Paar jedoch genauer, sieht man dass sich Bild (S,R) und Spiegelbild (R,S) zur Deckung bringen lassen. Beide Moleküle sind daher identisch !
Das 2R,3S-Isomer von Weinsäure ist achiral und deswegen nicht optisch aktiv, obwohl es zwei Chiralitätszentren enthält. Eine Verbindung, die zwei (oder mehr) Chiralitätszentren enthält, aber deckungsgleich mit ihrem Spiegelbild ist, bezeichnet man als Mesoverbindung. Alle Mesoverbindungen besitzen eine Spiegelebene, die das eine Chiralitätszentrum auf das andere abbildet.
Es sollte nochmals herausgestellt werden, dass es nur zwei Kriterien für Chiralität gibt: 1) Kann das Molekül mit seinem Spiegelbild zur Deckung gebracht werden? Wenn nicht, ist es chiral, oder 2) Enthaltet das Moleküle eine Symmetriezentrum oder eine Symmetrieebene? Wenn nicht, ist es chiral.
Noch ein paar Beispiele :
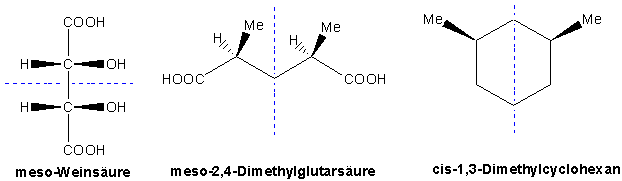
Diastereomere besitzen unterschiedliche relative Konfigurationen. Die relative Konfiguration ist die Beziehung zwischen der absoluten Konfiguration (oder der Händigkeit) der zwei enthaltenen Chiralitätszentren.
z.B.
Man kann die relative Konfiguration mit Hilfe der Erythro/Threo Nomenklatur bezeichnen.
Es muss sich um ein Molekül handeln, das zwei gleich substituierte (also, zwei identische Substituenten) Chiralitätszentren enthält. Das Molekül soll in einer Fischer Projektion geschrieben werden :
z.B.
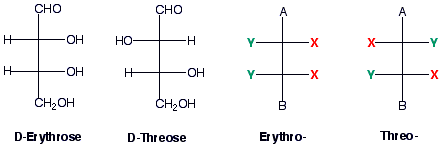
Die erythro/threo Nomenklatur ist oftmals nicht geeignet, weil viele Diastereomere diese Kriterien nicht erfüllen. In solchen Fällen kann die relative Konfiguration nur mit der R/S Nomenklatur angegeben werden.
Welche strukturelle Vielfalt können wir bei einer Verbindung mit drei Chiralitätszentren erwarten ? Dieses Problem können wir wieder durch Permutieren der verschiedenen Möglichkeiten lösen. Kennzeichen wir die drei Zentren nacheinander R oder S, ergeben sich die folgenden möglichen Stereoisomere :
| R-R-R | R-R-S | R-S-R | S-R-R | R-S-S | S-R-S | S-S-R | S-S-S |
|---|
also, insgesamt acht Stereoisomere. Sie lassen sich zu folgenden vier Enantiomerenpaaren ordnen :
| Bild | R-R-R | R-R-S | R-S-S | R-S-R |
| Spiegelbild | S-S-S | S-S-R | S-R-R | S-R-S |
Die Zahl der möglichen Stereoisomeren nimmt ab, wenn Mesoformen und gewisse cyclische Systeme auftreten. Allgemein gilt, dass eine Verbindung mit n Chiralitätszentren maximal 2n Stereoisomere haben kann. Bei grösseren Systemen ergeben sich, natürlich, phantastische strukturelle Möglichkeiten.
Oftmals entsteht bei der Bildung eines chiralen Moleküls aus achiralen Ausgangsstoffen ein racemisches Gemisch. Es hebt sich nun die Frage, wie man reine Enantiomere einer chiralen Verbindung erhält.
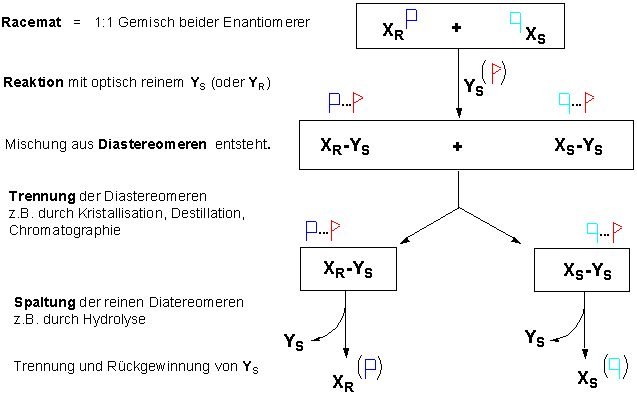
Eine mögliche Methode ist die, vom Racemat auszugehen und die Enantiomere voneinander zu trennen. Diesen Prozess bezeichnet man als Racematspaltung.
Aber Enantiomere besitzen identische physikalische und chemische Eigenschaften, mit Ausnahme ihrer Wechselwirkung zu anderen chiralen Molekülen und Phenomänen. Diastereomere besitzen dagegen unterschiedliche physikalische und chemische Eigenschaften und lassen sich durch fraktionierende Kristillation, Destillation oder Chromatographie trennen.
So ergäbe beispielsweise die Reaktion des Racemats XR,S mit einer optisch aktiven, chiralen Verbindung YS zwei optisch aktive Diastereomere :
Unten sehen wir, wie Naproxen (eine sogennante "Non-Steroidal-Anti-Inflammatory Drug", wie. z.B. Ibuprofen) auf diese Weise in die Enantiomere zerlegt wird. Man behandelt das Racemat zunächst mit einem Glucose-Derivat (welches aus der Natur in optisch reiner Form gewonnen werden kann), wobei die beiden diastereomeren Salze entstehen :
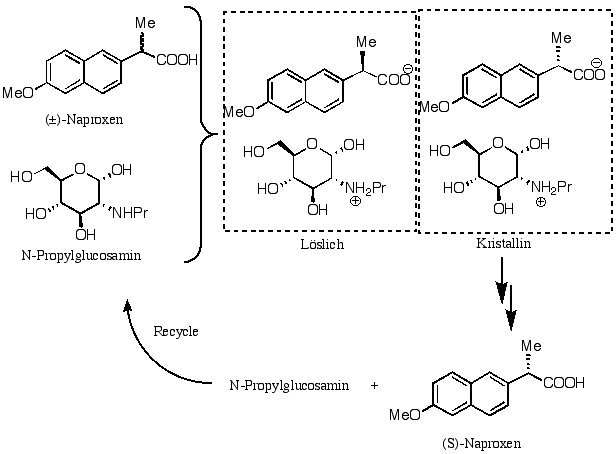
Das (S)-Isomer kristallisiert beim Stehenlassen als Salz mit dem Amin aus und kann von der Mutterlauge mit dem (R)-Isomer abfiltriert werden. Beim Behandeln des Salzes mit wässeriger Carbonatlösung kann das Amin extrahiert werden und (S)-Naproxen wird als Natriumsalz kristallisiert.
Die Spaltung eines racemischen Amins mit einer enantiomerenreinen Säure ist auch möglich. Es gibt viele andere Möglichkeiten, wie man die Bildung von Diastereomeren zur Racematspaltung benutzen kann.
Wir haben gerade bemerkt, dass Diastereomere unterschiedliche physikalische Eigenschaften besitzen, und sich durch fraktionierende Kristillation, Destillation oder Chromatographie trennen lassen. Sie besitzen auch unterschiedliche chemische Eigenschaften, was unterschiedliche Reaktivitäten gegenüber (chiralen) Reagenzien bedeuten kann oder nach einer Reaktion zu unterschiedlichen Produkten führen kann.
Wir haben auch bemerkt, dass Enantiomere mit Ausnahme ihrer Wechselwirkung zu anderen chiralen Molekülen und Phenomänen identische physikalische und chemische Eigenschaften besitzen. Fast alle chirale Biomoleküle kommen in optisch reinen Formen vor. Dass heisst, enantiomere Moleküle können in der Natur (im Vergleich zu ihren Eigenschaften im Labor) unterschiedliche Eigenschaften vorweisen .
z.B.:
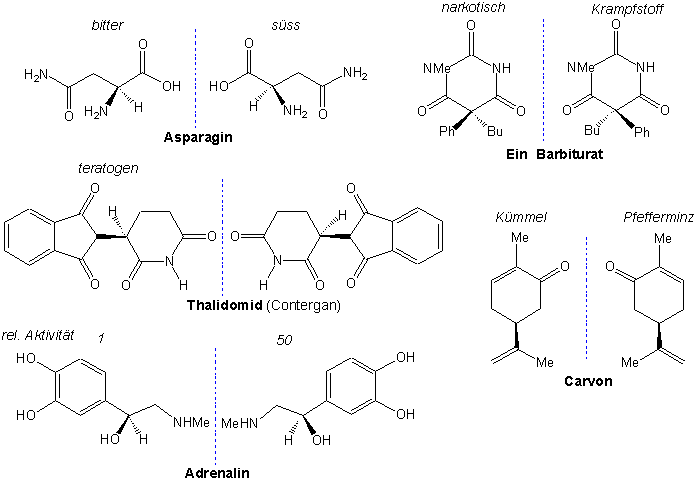
Warum haben solche Stereoisomere unterschiedliche biologische Eigenschaften ? Um ihren biologischen Effekt auszuüben müssen die Moleküle mit anderen Biomoleküle (Proteinen oder DNA, z.B.) Komplexe bilden. Die Biomoleküle sind chiral ! Ein chirales Molekül muss zu seinem spezifischen Rezeptor passen (wie eine Hand zu einem Handschuhe !). Zum Beispiel können wir in der folgenden Abbildung eine spezifische Wechselwirkung zwischen dem Rezeptor für Adrenalin und den zwei Adrenalin Stereoisomeren darstellen :
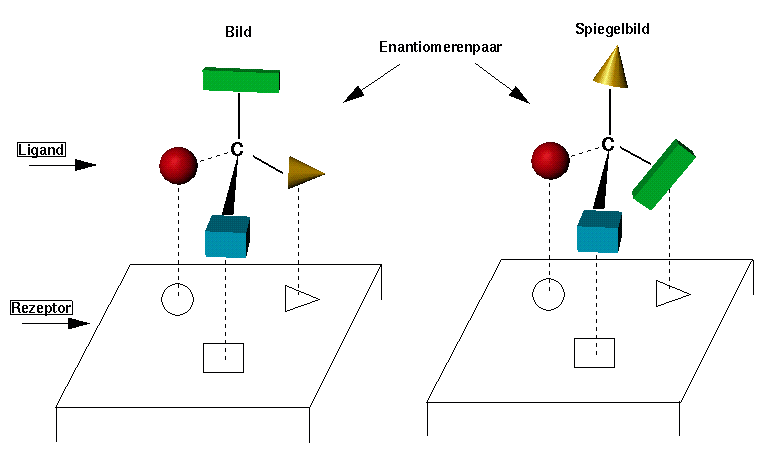
Nur einer der zwei Diastereomeren-Komplexe ist stabil, weil nur in einem Komplex jeder Substituent genau zu seiner Bindungsstelle passt.
| Übungsbeispiele zum Thema Stereochemie im Internet |
|---|
