H. J. Hansen
Research Interests:
The research interests of H.-J. Hansen may be described with physical organic chemistry in its broadest sense. The active research fields are Organic reaction mechanisms, organic photochemistry, transition metal chemistry, homogeneous asymmetric catalysis, high-pressure and high-temperature chemistry. They concentrated on the development of heptalene chemistry with respect to the synthesis of new thermo- and photochromic systems, of new ligand systems for asymmetric induction in transition-metal-catalyzed transformations, and of new benzo[d]heptalenes which would open new ways to the class of biologically active colchinoids. Further interest is focused on chirality transfer in excited states as well as on reactions in super-critical media such as super-critical carbon dioxide.
Over the past five years, the main research activities concentrated on the development of azulene and heptalene chemistry as well as on kinetic resolution of chiral substrates in the excited state by asymmetric sensitization. Some work has also been performed on high-temperature rearrangements by DGPTI (dynamic gas phase thermo-isorization).
Selected Publications:
A. J. Rippert, H. -J. Hansen,
'Ru-Catalyzed heptalene Formation from Azulenes and Dimethyl Acetylenedicarboxylate'
Helv. Chim. Acta 1992, 75, 2219.
R. -A. Fallahpour, H. -J. Hansen,
Formation of dimethyltricyclo[6.2.2.01,7]dodeca-2,4,6,9,11-pentaene-9,10-dicarboxylates from azulenes and dimethyl acetylenedicarboxylate under high pressure and thermal rearrangement of the tricyclic compounds into heptalene-1,2-dicarboxylates
High Pressure Res. 1992, 11, 125.
Y. Chen, R. W. Kunz, P. Uebelhart, R. H. Weber, H. -J. Hansen,
Thermal Reaction of Highly Alkylated Azulenes with Dimethyl Acetylene-dicarboxylate: HOMO (Azulene) vs. SHOMO (Azulene) Control in the Primary Thermal Addition Step
Helv. Chim. Acta 1992, 75, 2447.
A. J. Rippert, A. Linden, H. -J. Hansen,
Cationic RhI Complexes of Azulenes and their Catalytic Activity on the Formation of Heptalene-1,2-dicarboxylates from Dimethyl Acetylenedicar-boxylate and Azulenes
Helv. Chim. Acta 1993, 76, 2876.
A.
Magnussen, P. Uebelhart and H. -J. Hansen,
Thermal reaction of azulene-1-carboaldehydes with dimethyl acetylenedicarboxylate
Helv. Chim. Acta 1993, 76, 2887.
H. Heimgartner, H. -J. Hansen,
Struktur und Mechanismus in der Organischen Chemie im Spiegel von Helvetica Chimica Acta
Helv. Chim. Acta 1992, 75, 359.
H.
Heimgartner, H. -J. Hansen,
Organische Photochemie im Spiegel von Helvetica Chimica Acta
Helv. Chim. Acta 1993, 76, 1027.
A. A. S. Briquet, P. Uebelhart, H.-J. Hansen,
Double-Bond Shifts in [4n]Annulenes as New Principle for Molecular Switches: First Results with Dimethyl Heptalene-1,2- and -4,5-dicarboxylates
Helv. Chim. Acta 1996, 79, 2282-2315
S. El Houar, H.-J. Hansen,
New Heptalenes Substituted with Extended pi-Systems
Helv. Chim. Acta 1997, 80, 253-266
K. Abou-Hadeed, H.-J. Hansen,
An 'One-Pot' Anellation Method for the Transformation of Heptalene-4,5-dicarboxylates into Benzo[a]heptalenes
Helv. Chim. Acta 1997, 80, 2535-2564
M. Lutz, A. Linden, K. Abou-Hadeed, H.-J. Hansen,
Formation of 3-Sulfonyl Substituted Benzo[a]heptalene-2,4-diols from Heptalene-1,2-dicarboxylates and Lithiomethyl Phenyl Sulfones
Helv. Chim. Acta 1999, 82, 372-388
P. Uebelhart, A. Linden, H.-J. Hansen, Y.A. Ustynyuk, O.A. Trifanova, N.G. Akhmedov, V.I. Mstislawsky,
Tetramethylheptalenes and Their Tricarbonylchromium Complexes: Synthesis, Structures, and Thermal Rearrangements
Helv. Chim. Acta 1999, 82, 1930-1959
A. Linden, M. Meyer, P. Mohler, A.J. Rippert, H.-J. Hansen,
A Detailed Investigation of the Reaction of 5,9-Diphenylbenz[a]azulene with Dialkyl Acetylenedicarboxylates Leading to Dialkyl 8,12-Diphenylbenzo[a]heptalene-6,7-dicarboxylates
Helv. Chim. Acta 1999, 82, 2274-2315
A. Linden, M. Meyer, P. Mohler, A.J. Rippert, H.-J. Hansen,
An Anatomy of the Reaction of 5,9-Diphenylbenz[a]azulene with Dialkyl Acetylendicarboxylates Leading to Dialkyl 8,12-Diphenylbenzo[a]heptalene-6,7-dicarboxylates
Helv. Chim. Acta, 1999, 82, 2274.
P. Mohler, A.J. Rippert, H.-J. Hansen,
Synthesis of (P)- and (M)-6,7-Bis[(diphenylphosphanyl)methyl]-8,12-diphenylbenzo[a]heptalenes-Potential Ligands for Homogeneous Asymmetric Catalysis
Helv. Chim. Acta 2000, 83, 258-277
M. Nagel, H.-J. Hansen,
Synthesis of Polyalkylphenyl Propiolates and Their Flash Vacuum Pyrolysis into Polyalkylcyclohepta[b]furan-2(2H)-ones
Helv. Chim. Acta 2000, 83, 1022-1048
A.J. Rippert, A. Linden, H.-J. Hansen,
Formation of Diastereomerically Pure Oxazaphospholes and Their Reaction to Chiral Borylphosphanes
Helv. Chim. Acta, 2000, 82, 311.
V. Lellek, H.-J. Hansen,
Surprising Formation of Highly Substituted Azulenes on Thermolysis of 4,6,5,7,8-Pentamethylcyclohepta[b]furan-2(2H)-one and Heptalene Formation with the New Azulenes
Helv. Chim. Acta 2001, 84, 1712-1736
V. Lellek, H.-J. Hansen,
Unexpected Thermal Transformation of Aryl 3-Arylprop-2-ynoates- Formation of 3-(Diarylmethylidene)-2,3-dihydrofuran-2-ones
Helv. Chim. Acta 2001, 84, 3548-3580
Azulene and Heptalene Chemistry H.-J. Hansen




Azulenes are of interest for us as precursors for heptalene-4,5-dicarboxylates, which are formed on thermal reaction of azulenes with acetylenedicarboxylates (Hafner’s heptalene synthesis from 1976; for a review, see: Bull. Chem. Soc., Jpn., 1988, 61, 155). In our earlier work, we established the mechanism of this interesting ring-enlargement reaction of azulenes (see, e.g., Helv. Chim. Acta 1995, 78, 1933). The thermal and acid-catalyzed reaction of azulenes with acetylenedicarboxylates is fascinating with respect to the multitude of different reaction channels which can be entered (Scheme 1). Most interesting are the findings of Y. Chen (Ph.D. thesis, University of Zurich, Zurich, 1993) and E. Lehto (Ph.D. thesis, University of Zurich, Zurich, 1997) on the Et2AlCl -catalyzed reactions of azulenes with ADR. The primary intermediates of type C (Scheme 1), which are formed almost quantitatively, undergo an instantaneous cyclodimerization > under the influence of Et2AlCl. Summarizing examples are given in Scheme 2. All three cyclodimers of C show again their own purely thermal and acid-catalyzed chemistry. Other astonishing observations were made by us when we followed a published procedure for the oxidation of 1-methylazulenes with DDQ to the corresponding azulene-1-carbaldehydes (see: Okajima, Kurokova, Chem. Lett. 1997, 69). Indeed, the aldehydes are formed as main products. However, in yields up to 10% DDQ enters as reactant into the electron-transfer game with the azulenes (Scheme 3). The unexpected structure of G has been solved by X-ray crystal-structure analysis. We also think that we know the mechanism of its formation (publications for Helv. Chim. Acta in preparation). Over the last years, we have developed the most flexible synthesis of highly substituted azulenes starting with phenols. It is based on a combination of Trahanowsky’s flash vacuum pyrolysis (FVP) of aryl propiolates and Nozoe’s azulene synthesis by thermal reaction of cyclohepta[c]furan-2(2H)-ones and enol ethers or enamines (Scheme 4). However, as mentioned above, our azulene chemistry is mainly dedicated to the synthesis of heptalene-4,5-dicarboxylates. Heptalenes, as sigma-bridged [12]annulenes, represent a fascinating class of compounds, which exist, provided appropriate substitution, in two pi-bond states, describing constitutionally isomeric structures (Scheme 5). On the other hand, the heptalene skeleton is found as tetrahydrobenzo[a]heptalene structure in an important class of biologically highly active natural products, i.e., colchicine and its naturally occurring variants. Therefore, our momentary and final research interest is bipartite in a most thrilling way. On one hand, we are working in the field of material science and on the other hand, we are heavily involved in biological chemistry with a new synthetic concept (chemists à la mode normally call a synthetic concept "a strategy") for the synthesis of colchicinoids. In contrast to established syntheses, we construct ring A on the B—C arrangement of heptalenes (Scheme 5).
Colchicinoid Chemistry H.-J. Hansen



All of the so far known syntheses of colchicine and its derivatives follow synthetic pathways with precursors containing an intact aromatic ring A subunit. This leads to the two synthetic concepts A—B + C and A—C + B, where ring C is constructed on a ring A—B arrangement or where B is formed on an A—C precursor structure. Heptalene-4,5-dicarboxylates or their DBS (double-bond shifted) forms, the heptalene-1,2-dicarboxylates, represent a ring B—C construction, which contains already 4 C-atoms of the aromatic ring A. Our original idea was the transformation of the two EMe groups into ethynyl groups (or substituted ethynyl groups), so that a Bergman cyclization should be possible. In principle, this concept is practicable (see: K. Abou-Hadeed, Chimia 2000, 54, 763). However, the yields of the benzo[a]heptalenes by this route are too low (for another possibility, see: Helv. Chim Acta 2002, 85. fasc. 4). By chance, working on the "Bergman" concept, we found a very attractive and unprecedented transformation of the vicinal ester groups of the heptalenes under the action of lithiated methyl sulfones and excess of butyl lithium (BuLi) in a "one-pot" reaction into benzo[a]heptalene-2,4-diols (Scheme 6; see: Helv. Chim. Acta 1997, 80, 2535), the yields of which had been substantially improved in the course of our investigations (see: Helv. Chim. Acta 1999, 82, 372 and ibid. 2000, 83, 2383). This absolutely new transformation of maleic-ester substructures into resorcinols is not limited to heptalenedicarboxylates, but can also be realized with disubstituted maleic anhydrides themselves (see: K. Abou-Hadeed, Chimia 2000, 54, 760), which deliver under our controlled conditions 5,6-disubstituted 4-methylresorcinols. Despite the fact that we learned how to transform the 3-sulfonylated benzo[a]heptaleno-2,4-diols into 2,3,4-trimethoxybenzo[a] heptalenes (see: Chimia 2001, 55, 621), one has to recognize that the three MeO groups are in the "unnatural" 2,3,4-position. However, in the meantime we found a way how to force the originally formed two hydroxy groups in the 1,3-position with the conditio sine qua non that C(4) will carry, at least, a Me group (Scheme 7). Nevertheless, we master now this new C1-carrier reaction under rearrangement, whereby the first carrier group (XSO2) is lost already in the course of the benzo-anellation step as XSO2— and the second one can be removed reductively. Therefore, 1,2,3-trimethoxy-4-benzo[a]heptalenes are now available (see: Chimia 2001, 55, 641). In separate experiments, we have already realized the introduction of an AcNH group at C(7) (Scheme 8) as well as a bromo substituent at C(9) of the final benzo[a]heptalene structures (Scheme 8). One disadvantage of the described procedure of benzo[a]anellation of heptalenes might be the strongly basic conditions that have to be applied (BuLi in excess). Therefore, we have been looking for a second benzo[a]anellation process of heptalenes which also utilizes the present C-atoms of the vicinal heptalenedicarboxylates, but takes place under more or less neutral conditions. The fundamental reactions of this new synthesis of benzo[a]heptalenes, on which we reported only briefly so far (see Introductory Part, in: Helv. Chim. Acta 2000, 83, 2383), are displayed in Scheme 9. The synthesis of the heptaleno[1,2-c]furans, which we published earlier (see: Helv. Chim. Acta 1995, 78, 1437), has meanwhile been improved substantially. The heptaleno[1,2-c]furans undergo Diels-Alder addition reactions with all types of dienophiles. Most promising are the reactions with alpha-acetoxy-acrylonitrile or with (E)-1,2-di(phenylsulfonyl)-ethene. The momentary "state-of-the-art" of this second benzo[a]anellation procedure is also shown in Scheme 9. We are confident and without doubt that we will reach our aim, i.e., the establishment of a variable synthesis of colchicinoids, based on the construction principle B—C + A, which will allow much more flexibility in the variation of substituents.
Bis-pi-substituted heptalenes as new possible components formolecular switches and data storage systems H.-J. Hansen

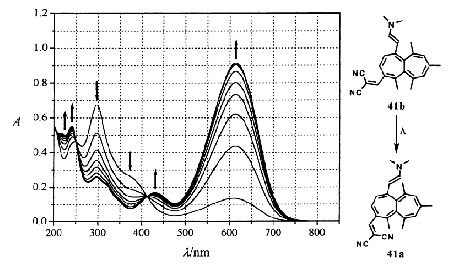
Our fundamental ideas of appropriately substituted heptalenes as new types of thermo- and photochromic systems have already been published (see: Helv. Chim. Acta 1996, 79, 2282 and ibid. 1997, 80, 1997), and a patent has also been filed on this matter by our University. Our latest results have been presented by TL at the conference: "Chemistry and Physics of Multifunctional Materials" at Acquafredda di Maratea (Italy), September 2001. We restrict in this report our considerations on the new horizons that we opened with our investigations. Almost all established thermo- and photochromic systems are working by interconversion of two completely different chemical states with corresponding different excited states that can easily be recorded. The states are reached by reversible ring opening and ring closure of extended pi-systems of merocyanine-type structures, i.e., structures equipped with pi-donor and pi-acceptor groups. The global situation can be described by the chemical bonding equation: p D s + (p-1). With respect to the accompanying longest-wavelength band shift one speaks of positive or negative thermo- and photochromism. We call these systems, which are based on the batho- and hypsochromism, type I thermo- and photochromic systems. In heptalenes, the two DBS isomers (cf. Scheme 5) represent the same chromophoric system, however, with different relative positions of substituents with respect to the pi-bond perimeter. In the on-state form (Scheme 5) one observes a conjugative interaction of the two pi-substituents, which are placed in 1,4-relation to each other. This "through-conjugation" causes a dramatic hyperchromic effect on the longest-wavelength absorption band. Irradiation into this absorption band generates efficiently the DBS isomer, i.e., the "off-state" form with an interruption of the direct conjugative interaction of the two pi-substituents. This effects a massive break-down of the intensity of the longest-wavelength absorption band (hypochromic effect) of the DBS isomer. Nevertheless, it is a heptalene of the same type. Therefore, it possesses also an absorption band almost at the same position as the "on-state" isomer (see the UV/VIS spectra displayed in: Helv. Chim. Acta 1997, 80, 253). We call these systems, which follow the simple bonding equation: p D p’ and are based on hyper- and hypochromism, type II thermo- and photochromic systems. Extensive DFT calculations show that the transition state of the thermal DBS process as well as the lowest lying singlet state (S1) are still twisted as their ground state (S0), but to a much smaller extent. However, more important is the fact that both states (TS and S1) exhibit almost perfect bond length equalization. In other words, the S1 state of the heptalenes can smoothly relax to the ground state, thereby causing DBS. Indeed, we found already very early that the thermal as well as the photochemical DBS process takes place with retention of configuration of the inherently chiral heptalene core (see: Helv. Chim. Acta 1985, 68, 429). We resigned in this part to discuss the synthetic efforts to get the bis-pi-substituted heptalenes. It’s indeed a chemistry in itself. The fundamental chemistry has been developed by Sarah Maillefer-El Houar (see: Helv. Chim. Acta 1997, 80, 253), extended by Jianfeng Song (see: Helv. Chim. Acta 1999, 82, 1690 and 2260), and brought to full blossoming by Thomas Landmesser (see also: Chimia 2001, 55, 624). Two figures, taken from the Ph.D. thesis of TL, may document our success (see: Figure of one of our most developed thermo- and photochromic systems based on heptalenes). Figure: UV/VIS Absorption Spectra of Heptalenes 41a/41b In MeCN in the Course of Irradiation (left) and Thermal Recovery (right; the arrows indicate the decrease and increase of the corresponding band intensities). Irradiation at 600 nm in intervals of 10 s. Thermal recovery at ambient temperature; time intervals: 0, 15, 30, 45, 60, 90, 150, and 210 min.
Homoheptalene Chemistry
H.-J. Hansen
Good 20 years ago, Scott et al. published a synthesis of homoheptalenedicarboxylates starting with homoazulene and ADM (see: J. Am. Chem. Soc. 1981, 103 and ibid. 1982, 104, 3530). We resumed this chemistry recently (see: Helv. Chim. Acta 2001, 84, 1017) and could extend the views of Scott et al. At the moment, we try to transfer our experiences in heptalene chemistry to homoheptalenes (running Ph.D. thesis work of Georg Rüedi), which is a much more difficult task than we thought at the beginning. Nevertheless, DFT calculations on the DBS process of homoheptalene itself led to an activation energy of 11 kcal/mol, whereby the transition state shows C2v symmetry. We hope that appropriately substituted homoheptalenes may represent rapid molecular switches at r.t.
Kinetic Optical Resolution in the Excited State
H.-J. Hansen

So far, we have not published any results of this project that has been followed over the past 10 years with postdoctoral coworkers of the Technical University or Prague. Therefore, we will discuss here only the general principle. It is well-established that conjugated trienes can be trans,cis-isomerized by triplet sensitization (see, e.g., W. G. Dauben et al., J. Am. Chem. Soc. 1991, 113, 8367 and lit. cited there). Working with optically active sensitizers such as the 1,1’-bisanthracenes and racemic cis- or trans-configured trienes such as those in Scheme 10 should lead to diastereoisomeric encounter-complexes with different triplet-energy transfer rates. If these transfer rates differ sufficiently (k(Rsens,Rtriene)/k(Rsens, Striene) pi 1), the sensitized trans, cis-isomerization must be accompanied by optical resolution of the geometrical isomers. With the shown systems, we have observed secured ee values of 5—8%. That’s not very much. However, a number of established asymmetrically catalyzed reactions started with similar figures. At the moment, we are synthesizing new types of sensitizers on anthracene basis (running Ph.D. thesis work of Christian Orellano).
Kinetic Optical Resolution in the Excited State
H.-J. Hansen
So far, we have not published any results of this project that has been followed over the past 10 years with postdoctoral coworkers of the Technical University or Prague. Therefore, we will discuss here only the general principle. It is well-established that conjugated trienes can be trans,cis-isomerized by triplet sensitization (see, e.g., W. G. Dauben et al., J. Am. Chem. Soc. 1991, 113, 8367 and lit. cited there).
Working with optically active sensitizers such as the 1,1’-bisanthracenes and racemic cis- or trans-configured trienes such as those in Scheme 10 should lead to diastereoisomeric encounter-complexes with different triplet-energy transfer rates. If these transfer rates differ sufficiently (k(Rsens,Rtriene)/k(Rsens, Striene) pi 1), the sensitized trans, cis-isomerization must be accompanied by optical resolution of the geometrical isomers. With the shown systems, we have observed secured ee values of 5—8%. That’s not very much.
However, a number of established asymmetrically catalyzed reactions started with similar figures. At the moment, we are synthesizing new types of sensitizers on anthracene basis (running Ph.D. thesis work of Christian Orellano).
Perfectly Working Repeatable Ring-Enlargement Reactions at 650°C
H.-J. Hansen
This work has been performed in cooperation with Givaudan/Dübendorf by Matthias Nagel in the course of his Ph.D. thesis work (Neuartige Ringerweiterungsreaktionen durch Dynamische Gasphase-Thermo-Isomerisierung zur Synthese makrocyclischer Alkanone, University of Zurich, Zurich 2001). In the course of investigations aimed at a technical synthesis of campholenic aldehyde, it was found that n-membered 1-vinylcycloalkanols rearrange at 650° under DGPTI conditions in high yields into the corresponding (n+2)-membered cycloalkanones, which, in turn, can be reacted with vinylcerium reagents and then thermally ring-enlarged again (Scheme 11; for details, see: Synlett 2002, 275 and 280). This work is going to be continued with other substrates.
We suppose that this high-temperature ring enlargement of medium-sized and large rings is so successful, because homolytric bond cleavage and bond formation take place on a time scale where conformational changes in the formed biradicals are too slow to compete effectively with the bond-forming event.
Tricarbonylchromium Complexes of Heptalene
H.-J. Hansen
We started this project several years ago in cooperation with Yuri Ustynyuk and his group at Moscow State University. IWe could identify two different haptotropic rearrangements in tricarbonyl(heptalene)chromium complexes characterized by an intramolecular intra-ring switch of the Cr(CO)3 group, which is accompanied by DBS, and an inter-ring jump of the Cr(CO)3 group from one seven-membered ring to the other (for details, see: Helv. Chim. Acta 1999, 82, 1930). We have continued this work, specifically with respect to the formation of Cr(CO)3 complexes of heptalenedicarboxylates. As expected, the Cr(CO)3 complexes, where the Cr(CO)3 group sits at the alkyl substituted ring are thermodynamically favoured. (to be published, Helv. Chim. Acta).
Raman-Optical-Activity (ROA) Spectroscopy
H.-J. Hansen
This is a resumed cooperative research project, which can be traced back to my time as professor in Fribourg (see, e.g., H.-J. Hansen, H.-R. Sliwka, W. Hug, Helv. Chim. Acta 1979, 62, 1120; W. Hug, A. Kamatari, K. Srinivasan, H.-J. Hansen, H.-R. Sliwka, Chem. Phys. Letters 1980, 76, 469); but where the leadership lies exclusively in the hands of W. Hug. In the past years, a new second-generation ROA spectrometer has been constructed by Werner Hug at our Institute in Zurich, which is of much higher sensitivity than the first one in Fribourg, which had been destroyed there by a fire in March 1981. A wonderful example of the quality of spectra, recorded with the new ROA spectrometer, has been published in Helv. Chim. Acta last year (2001, 84, 1). The ROA spectrum of (+)-(R)-1-methyl-[4,5,6,7-2H4]indane, which we have synthesized recently, clarified an open question of our earlier ROA measurements of (R)- and (S)-1-methylindane and deuterated derivatives of them (see: loc. cit., above), which all showed a characteristic ROA couplet at about 1376 cm—1. The break-down of this couplet in the upper case demonstrated clearly that the discussed couplet has its origin in a coupled vibration of the methyl group at C(1) and the in-plane movement of the aromatic H-atoms. This cooperative work on ROA spectroscopy will be continued. Our most ambitious candidates are (R)- and (S)-[ 2H1], [2H2],[ 2H3]neopentane, the syntheses of which we are elaborating at the moment.
Allocolchicine Syntheses H.-J. Hansen
This work on the utilization of Fischer’s carbenoid chemistry is performed in cooperation with William D. Wulff (Michigian State University). A first part of this work has already been published (see: Org. Lett. 2001, 3, 2641). It represents the Ph.D. thesis work of Andrei Voroguchin who is always spending half a year at William’s laboratory and half a year at my laboratory.